To: Administrative File: CAG-00430N
From: Louis Jacques, MD
Director, Coverage and Analysis Group
Tamara Syrek Jensen, JD
Deputy Director, Coverage and Analysis Group
Jyme Schafer, MD, MPH
Director, Division of Medical and Surgical Services
Sarah Fulton, MHS
Lead Analyst, Division of Medical and Surgical Services
Lawrence Schott, MD, MS
Lead Medical Officer, Division of Medical and Surgical Services
JoAnna Baldwin, MS
Technical Advisor, Division of Medical and Surgical Services
Subject: Coverage Decision Memorandum for Transcatheter Aortic Valve Replacement (TAVR)
Date: May 1, 2012
I. Decision
The Centers for Medicare & Medicaid Services (CMS) covers transcatheter aortic valve replacement (TAVR) under Coverage with Evidence Development (CED) with the following conditions:
TAVR is covered for the treatment of symptomatic aortic valve stenosis when furnished according to an FDA approved indication and when all of the following conditions are met.
The procedure is furnished with a complete aortic valve and implantation system that has received FDA premarket approval (PMA) for that system’s FDA approved indication.
Two cardiac surgeons have independently examined the patient face-to-face and evaluated the patient’s suitability for open aortic valve replacement (AVR) surgery; and both surgeons have documented the rationale for their clinical judgment and the rationale is available to the heart team.
The patient (preoperatively and postoperatively) is under the care of a heart team: a cohesive, multi-disciplinary, team of medical professionals. The heart team concept embodies collaboration and dedication across medical specialties to offer optimal patient-centered care.
TAVR must be furnished in a hospital with the appropriate infrastructure that includes but is not limited to:
- On-site heart valve surgery program,
- Cardiac catheterization lab or hybrid operating room/catheterization lab equipped with a fixed radiographic imaging system with flat-panel fluoroscopy, offering quality imaging,
- Non-invasive imaging such as echocardiography, vascular ultrasound, computed tomography (CT) and magnetic resonance (MR),
- Sufficient space, in a sterile environment, to accommodate necessary equipment for cases with and without complications,
- Post-procedure intensive care facility with personnel experienced in managing patients who have undergone open-heart valve procedures,
- Appropriate volume requirements per the applicable qualifications below.
There are two sets of qualifications; the first set outlined below is for hospital programs and heart teams without previous TAVR experience and the second set is for those with TAVR experience.
Qualifications to begin a TAVR program for hospitals without TAVR experience:
The hospital program must have the following:
- ≥ 50 total AVRs in the previous year prior to TAVR, including ≥ 10 high-risk patients, and;
- ≥ 2 physicians with cardiac surgery privileges, and;
- ≥ 1000 catheterizations per year, including ≥ 400 percutaneous coronary interventions (PCIs) per year.
Qualifications to begin a TAVR program for heart teams without TAVR experience:
The heart team must include:
- Cardiovascular surgeon with:
- ≥ 100 career AVRs including 10 high-risk patients; or
- ≥ 25 AVRs in one year; or
- ≥ 50 AVRs in 2 years; and which include at least 20 AVRs in the last year prior to TAVR initiation; and
- Interventional cardiologist with:
- Professional experience with 100 structural heart disease procedures lifetime; or;
- 30 left-sided structural procedures per year of which 60% should be balloon aortic valvuloplasty (BAV). Atrial septal defect and patent foramen ovale closure are not considered left-sided procedures; and
- Additional members of the heart team such as echocardiographers, imaging specialists, heart failure specialists, cardiac anesthesiologists, intensivists, nurses, and social workers; and
- Device-specific training as required by the manufacturer.
Qualifications for hospital programs with TAVR experience:
The hospital program must maintain the following:
- ≥ 20 AVRs per year or ≥ 40 AVRs every 2 years; and
- ≥ 2 physicians with cardiac surgery privileges; and
- ≥ 1000 catheterizations per year, including ≥ 400 percutaneous coronary interventions (PCIs) per year.
Qualifications for heart teams with TAVR experience:
The heart team must include:
- A cardiovascular surgeon and an interventional cardiologist whose combined experience maintains the following:
- ≥ 20 TAVR procedures in the prior year, or;
- ≥ 40 TAVR procedures in the prior 2 years; and
- Additional members of the heart team such as echocardiographers, imaging specialists, heart failure specialists, cardiac anesthesiologists, intensivists, nurses, and social workers.
The heart team’s interventional cardiologist(s) and cardiac surgeon(s) must jointly participate in the intra-operative technical aspects of TAVR.
The heart team and hospital are participating in a prospective, national, audited registry that: 1) consecutively enrolls TAVR patients; 2) accepts all manufactured devices; 3) follows the patient for at least one year; and 4) complies with relevant regulations relating to protecting human research subjects, including 45 CFR Part 46 and 21 CFR Parts 50 & 56. The following outcomes must be tracked by the registry; and the registry must be designed to permit identification and analysis of patient, practitioner and facility level variables that predict each of these outcomes:
- Stroke;
- All cause mortality;
- Transient Ischemic Attacks (TIAs);
- Major vascular events;
- Acute kidney injury;
- Repeat aortic valve procedures;
- Quality of Life (QoL).
The registry should collect all data necessary and have a written executable analysis plan in place to address the following questions (to appropriately address some questions, Medicare claims or other outside data may be necessary):
- When performed outside a controlled clinical study, how do outcomes and adverse events compare to the pivotal clinical studies?
- How do outcomes and adverse events in subpopulations compare to patients in the pivotal clinical studies?
- What is the long term ( ≥ 5 year) durability of the device?
- What are the long term ( ≥ 5 year) outcomes and adverse events?
- How do the demographics of registry patients compare to the pivotal studies?
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
TAVR is covered for uses that are not expressly listed as an FDA approved indication when performed within a clinical study that fulfills all of the following.
The heart team’s interventional cardiologist(s) and cardiac surgeon(s) must jointly participate in the intra-operative technical aspects of TAVR.
- As a fully-described, written part of its protocol, the clinical research study must critically evaluate not only each patient’s quality of life pre- and post-TAVR (minimum of 1 year), but must also address at least one of the following questions:
- What is the incidence of stroke?
- What is the rate of all cause mortality?
- What is the incidence of transient ischemic attacks (TIAs)?
- What is the incidence of major vascular events?
- What is the incidence of acute kidney injury?
- What is the incidence of repeat aortic valve procedures?
- The clinical study must adhere to the following standards of scientific integrity and relevance to the Medicare population:
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants’ health outcomes.
- The research study is well supported by available scientific and medical information or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it also must be in compliance with 21 CFR Parts 50 and 56. In particular, the informed consent includes a straightforward explanation of the reported increased risks of stroke and vascular complications that have been published for TAVR.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed as Medicare coverage requirements.
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject.
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria affect enrollment of these populations, and a plan for the retention and reporting of said populations on the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
- The principal investigator must submit the complete study protocol, identify the relevant CMS research
question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Re: TAVR CED
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
- TAVR is not covered for patients in whom existing co-morbidities would preclude the expected benefit from correction of the aortic stenosis.
II. Background
Throughout this document we use numerous acronyms, some of which are not defined as they are presented in direct quotations. Please find below a list of these acronyms and corresponding full terminology.
AATS – American Association for Thoracic Surgery
ACC – American College of Cardiology
ACCF – American College of Cardiology Foundation
AS – Aortic Stenosis
AVR – Aortic Valve Replacement
CK – Creatine Kinase
COPD - Chronic Obstructive Pulmonary Disease
CV – Cardiovascular
EVAR – Endovascular Aneurysm Repair
LVEF – Left Ventricular Ejection Fraction
MB – Myocardial Band
MI – Myocardial Infarction
PAS – Post Approval Study
PCI – Percutaneous Coronary Intervention
PI – Primary Investigator
RCT – Randomized Controlled Trial
SAVR - Surgical Aortic Valve Replacement
SCAI – Society for Cardiovascular Angiography and Interventions
STS – Society of Thoracic Surgeons
TAVR – Transcatheter Aortic Valve Replacement
TAVI – Transcatheter Aortic Valve Implantation
TEVAR – Thoracic Endovascular Aortic Repair
WHO – World Health Organization
The published literature uses both TAVR and TAVI to refer to the subject of this review. Readers may consider these terms to be interchangeable for the purposes of this memorandum.
Aortic Stenosis
The most common valvular abnormality in the United States is aortic stenosis (AS), with an incidence of approximately five of every 10,000 adults (Dewey 2008). As our population ages, AS prevalence will continue to increase. Aortic valve disease exists as a continuum, and aortic valvular abnormalities are often seen in older individuals as demonstrated by the Cardiovascular Health Study in which 26% of participants, men and women over the age of 65, had a degree of aortic sclerosis (Carabello 2009). Aortic sclerosis, which is an irregular valve thickening with no obstruction to ventricular blood outflow, is associated with age, sex, hypertension, smoking, diabetes, and serum LDL and lipoprotein levels and may progress to AS. The natural history in adults involves a long latent period where both morbidity and mortality are low. The progression of aortic stenosis to serious outflow obstruction causing sickness and death can be estimated, but much variability exists in the rate of progression, and it is not possible to predict the rate of progression in an individual patient. After the long latent period, symptoms of angina, syncope or heart failure can develop. Symptomatic severe aortic stenosis carries a poor prognosis (Moat 2011). On average, the survival is two to three years after symptoms develop, with a high risk of sudden death (Bonow 2008).
The most common cause of aortic stenosis in adults is calcification of the valve. This calcification progresses from the base of the cusps to the leaflets, and eventually causes a reduction in both leaflet motion and the effective valve area. This calcific disease is similar to atherosclerosis. Rheumatic AS disease, related to valvular infection, is less common. In young adults, congenital valve malformations are the more common cause for AS. The first sign of AS may be a murmur, detected during auscultation of the chest. If a murmur is detected, echocardiography may be indicated. Echocardiographic objective measurements include aortic jet velocity, mean pressure gradient and valve area. However, no single objective laboratory value defines severity or is the primary determinant of the need for valve replacement. Some patients with severe AS are asymptomatic, whereas others with only moderate stenosis develop symptoms. Therefore, therapeutic decisions are based mostly on the presence or absence of symptoms. For asymptomatic AS patients, the 2008 ACC/AHA guidelines recommend frequent monitoring for symptoms (which may be subtle), as well as disease progression (Bonow 2008). When patients develop symptoms thought to be due to AS, surgery is recommended.
Aortic Valve Replacement
Surgical aortic valve replacement (AVR) has been the gold standard for treatment in adults with severe symptomatic aortic stenosis and well-defined treatment guidelines exist (Dewey 2008). Until recently, surgical AVR has been the only effective treatment. In patients selected for isolated valve repair, the perioperative risk is low. Perioperative mortality in the Society of Thoracic Surgeons (STS) database is 3.0% to 4.0% for isolated AVR and 5.5% to 6.8% for AVR and coronary artery bypass graft (CABG) (Bonow 2008). Studies have shown that even in octogenarians AVR operative mortality was about 5-6%, with five year survival of 64-77% (Filsoufi 2008; ElBardissi 2011). Outcomes can vary based on surgical volume (Bonow 2008). However, risk can be increased for some patients (Moat 2011).
Despite clear guidelines, excellent surgical outcomes, and high mortality of symptomatic valve disease, some patients do not receive necessary treatment. “Some patients with severe symptomatic aortic stenosis do not undergo aortic valve replacement despite demonstrated symptomatic and survival advantages and despite unequivocal guideline recommendations for surgical evaluation” (Bach 2009). Bach and colleagues estimate that one third of patients with severe AS are symptomatic but do not undergo surgical replacement, with the findings not limited to any specific practice environment. For many of these patients who were not operated on, objective ascertainment did not reject the possibility of surgery with apparent involvement of both physician and patient subjective decision-making. The conclusion has been drawn that some patients with severe symptomatic AS may be inappropriately denied access to potentially life-saving surgery without clear explanation (Bach 2009).
TAVR
Technologic advancements have allowed for the delivery of heart valves via catheter as an alternative to open surgical valve replacement. The first in man studies were performed in 2002, and as such, TAVR is a relatively new procedure. TAVR treats the stenotic heart valve by displacing and functionally replacing the native aortic valve with a bioprosthetic valve delivered on a catheter via a percutaneous transarterial approach through a peripheral artery (e.g., the femoral artery), a transaortic approach through a limited sternotomy, or a transapical approach through a limited lower thoracotomy. Two devices, the SAPIEN and the CoreValve prostheses, are currently under post-market surveillance in Europe. The valve delivery system for these devices is similar, but the final step of implantation differs. The SAPIEN valve is a balloon-expandable bioprosthesis, whereas the CoreValve represents a self-expandable nitinol frame bioprosthesis. Proper technique with either is crucial. Though these implanted valves have been in use outside of the United States and sovereign registries exist to ascertain patient outcomes, none except for Moat and colleagues all-inclusive registry (with now two year outcomes) in the United Kingdom have yet reported significant numbers of consecutively enrolled patients with long-term follow-up. This is of great importance as the valve is expected to last the life of the patient (Moat 2011).
Postoperative complications lead to patient suffering, as well as increased burden. Therefore, it is important to identify patients who are at increased risk for surgical complications to guide future treatment decisions. Historically, this was a decision based on personal experience of the surgeon. To help in this decision and to provide reliable and accurate information for patients, a number of risk scoring systems have been developed. Saxton and Velanovich (2011) noted “the usefulness of the available scoring systems for accurately predicting postoperative complications is quite variable among different patient populations, indications for surgery, and surgical procedures performed.” This situation exists in large part because, although many morbidity and mortality risk factors for these scores have been extensively analyzed, considerable uncertainty remains regarding which patients will actually experience adverse outcomes. This is especially true in the elderly. Foremost among factors that have undergone investigation are patient age, comorbidities, physical examination findings and laboratory values (Saxton and Velanovich 2011). Recently, there has also been interest in more abstract concepts such as frailty and quality of life as risk predictors. Frailty is used to define older adults with impaired resistance to stressors due to decline in physiologic reserve, and is felt by some to better reflect biologic age as opposed to chronologic age (Afilalo 2011). Ultimately, however, any procedure is a risk-benefit decision. It is a critical endeavor to accurately determine such risk-benefit information for patient decision-making and empowerment.
III. History of Medicare Coverage
Until this NCD, CMS had no national policy that addressed coverage of TAVR or, as it is also known, TAVI.
Benefit Category
For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act (the Act). TAVR falls under the benefit categories set forth in section §1861(b)(3) (inpatient hospital services), a Part A benefit under §1812(a)(1), and §1861(s)(1) (physician services), a Part B benefit. This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
Current Request
On September 22, 2011, we received a formal complete written request from The Society of Thoracic Surgeons and The American College of Cardiology, submitted jointly. The request, available at http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id257.pdf, notes that the clinical outcomes reported in the pivotal trial were achieved when specific criteria were met.
Thus, we are asked to establish national Medicare coverage for TAVR with conditions of coverage, specifically when the procedure is
- “Performed in a specialized heart center utilizing a modified conventional cardiac laboratory or hybrid operating room that contains the specialized equipment necessary for the procedure;
- Managed using a multidisciplinary team using planned approach to co-management decision making as well as technical insertion of the device;
- Reported on using a joint STS-ACC TVT Registry.”
The joint specialty society request also recommends that CMS “include as a condition of coverage mandatory reporting of the procedures in an STS-ACC Transcatheter Valvular Therapy (TVT) Registry which would include long term follow-up using CMS data.”
IV. Timeline of Recent Activities
| Date |
Action |
| September 28, 2011 |
CMS accepts formal request from the Society of Thoracic Surgeons (STS) and American College of Cardiology (ACC), and initiates this national coverage analysis for transcatheter aortic valve replacement. The initial 30-day public comment period begins. |
| October 28, 2011 | Initial 30-day public comment period closes. |
| February 2, 2012 | Proposed decision memorandum is posted, second 30-day public comment period begins. |
| March 3, 2012 | Second 30-day public comment period closes. |
V. FDA Status
On November 2, 2011 the Food and Drug Administration (FDA) approved the first TAVR device for marketing in the United States. The Edwards’ Sapien Transcatheter Heart Valve (THV) was approved “for transfemoral delivery in patients with severe symptomatic native aortic valve stenosis who have been determined by a cardiac surgeon to be inoperable for open aortic valve replacement and in whom existing co-morbidities would not preclude the expected benefit from correction of the aortic stenosis” (http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P100041).
FDA approval (http://www.accessdata.fda.gov/cdrh_docs/pdf10/P100041a.pdf) includes a statement recommending specific training and experience for practitioners to use the device, as well as continued clinical study and data submission to the STS ACC TVT Registry.
VI. General Methodological Principles
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public commenters sometimes cite the published clinical evidence and provide CMS with useful information. Public comments that provide information based on unpublished evidence, such as the results of individual practitioners or patients, are less rigorous and, therefore, less useful for making a coverage determination. CMS uses the initial comment period to inform its proposed decision. CMS responds in detail to the public comments that were received in response to the proposed decision when it issues the final decision memorandum.
VII. Evidence
A. Introduction
This presentation of evidence primarily focuses upon whether the pivotal PARTNER randomized controlled trials (RCTs) are adequate to draw conclusions about health outcomes for TAVR, as well as whether the body of evidence is generalizable to the Medicare population. The evidence CMS examines has as its focus health outcomes, i.e., the benefits and harms of a particular treatment. Key outcomes of interest to CMS are periprocedural and long-term risk of stroke or death, as well as health-related quality of life and function post-TAVR. Independently assessed, validated instruments are most heavily weighted.
We summarize the evidence relating to the treatment of symptomatic aortic stenosis with the transcatheter aortic valve which includes the FDA premarket application clinical trial that was divided into two groups (PARTNER A and PARTNER B) based upon patient selection criteria. Previous case series studies have also been reviewed, but add little to the conclusions of the PARTNER studies as they are either non-consecutive or small. In treatment of symptomatic aortic stenosis, the primary focus is reduction in symptoms (chest pain, shortness of breath, fatigue and weakness), cardiovascular events (heart failure, stroke, myocardial infarction and arrhythmia) and mortality (cardiovascular mortality), as well as improvement in QoL and function.
Study endpoints should be clearly defined a priori to both improve the quality of clinical research and so as to allow comparison between clinical trials. For TAVI clinical trials, a report was published proposing standardized consensus definitions for important clinical endpoints (Leon 2011). In this consensus document, the following outcomes were given clinically relevant definitions:
- Cardiovascular mortality,
- Myocardial infarction,
- Stroke,
- Bleeding,
- Acute kidney injury,
- Vascular access site and access-related complications,
- Potential failure modes of prosthetic valve dysfunction.
Functional outcome measures for aortic stenosis include the New York Heart Association (NYHA) classification (I-IV), the six minute walk test (6MWT), the fifty meter walk test, and the modified Rankin Scale (mRS). The NYHA classification is a subjective symptom measure. The six minute walk test is (as the name describes) a standardized approach, which can be effort dependent, and the 6MWT is similar to the fifty meter walk test. The mRS is a measure of stroke disability and reportedly provides a better impression of whether patients are able to look after themselves than activities of daily living (ADL) scores (Van Swieten 1988). The mRS has six classifications: 0 = no symptoms; 1 = no significant disability; 2 = slight disability; 3 = moderate disability; 4 = moderately severe disability, unable to walk without assistance, and unable to attend to own bodily needs without assistance; 5 = severe disability, including being bedridden, incontinent, and those requiring constant nursing care and attention.
Quality of life is important to Medicare beneficiaries and can weigh heavily in patients’ decision- making. Therefore, valid and reliable measurement is important to inform patients. Quality of life measures can be disease specific or general. Disease specific measures used in TAVR trials have included the Kansas City Cardiomyopathy Questionnaire (KCCQ), which is a 23-item questionnaire for assessment of disability and quality of life impairment due to congestive heart failure. Other heart failure assessments include the Minnesota Living with Heart Failure. Generic QoL assessments included the SF-36, SF-12, PROMISE, and the EuroQoL for population comparisons. There are advantages and disadvantages to each tool, and the end use can help with tool choice, i.e., disease specific to measure within the population, and generic for a broad population comparison. Physiologic measures such as hemodynamic measurement by echocardiography are also used but their relationship to clinical outcomes is less clear.
B. Discussion of Evidence
1. Questions:
The development of an assessment in support of Medicare coverage decisions is based on the same general question for almost all national coverage analyses (NCAs): "Is the evidence sufficient to conclude that the application of the item or service under study will improve health outcomes for Medicare patients?" For this NCD, the questions of interest are:
- Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for Medicare beneficiaries with severe symptomatic aortic stenosis who are not candidates for surgical aortic valve replacement?
- Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for high surgical risk Medicare beneficiaries with severe symptomatic aortic stenosis who are candidates for surgical aortic valve replacement?
If the answer to either or both of the questions above is positive, is the available evidence adequate to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from TAVR?
2. External Technology Assessment (TA)
CMS did not commission an external TA for this NCA, but an October 12, 2011 Belgian health technology assessment (Neyt 2011), an interventional procedure overview/review (NICE 2011), an interventional procedure guidance (NICE March 28, 2012) and a health technology assessment (California Technology Assessment Forum 2012) were identified which analyzed the PARTNER study.
Belgian Health Technology Assessment (2011)
Neyt M, Van Brabandt H, Van de Sande S, et al. Health technology assessment. Transcatheter aortic valve implantation (TAVI): a health technology assessment update. Health Technology Assessment (HTA) 2011 Brussels: Belgian Health Care Knowledge Centre (KCE). KCE Reports
163C. D/2011/10.273/48 Available online: http://kce.fgov.be/sites/default/files/page_documents/kce_163c_tavi_update.pdf
The 2011 Belgian Health Technology Assessment made the following key points about the PARTNER trial in general (as well as for both cohorts) and also provided critical analysis regarding particularly the PARTNER trial’s internal validity and physicians’ preferences:
General Remarks
- “TAVI is a highly invasive and challenging procedure addressing elderly people in poor general condition. The procedure takes on average over 4 hours (skin-to-skin time 2 to 3 hours). It involves prolonged general anaesthesia, the administration of contrast media, and trans-oesophageal echocardiography. It is complicated with hemorrhagic vascular adverse events in more than 50% of patients.
- Differentiating “patients who cannot undergo surgery” (PARTNER Cohort B) from "surgical high-risk patients” (Cohort A) essentially relies on the clinical feeling of the physicians involved.
- The treatment effect of TAVI may be overestimated in PARTNER because of methodological concerns and a potential impact of conflicts of interest. Long term outcomes related to a residual aortic regurgitation after TAVI, and the long term durability of the prosthesis remain unknown.”
PARTNER Cohort A
- “In patients with aortic stenosis who are at very high surgical risk, TAVI and surgery are associated with a similar mortality rate at 30 days and 1 year and produce similar improvements in cardiac symptoms.
- The above mentioned observation dissolves our initial safety concerns of TAVI, but the approximate doubling in the rate of stroke 1 year after TAVI (8.3%) compared to surgery (4.3%) remains a concern.
- The 30-day mortality rate of TAVI observed in Cohort A of the PARTNER trial is the lowest ever reported in a TAVI study although most of the participating centres had no previous experience with TAVI.”
PARTNER Cohort B
- “The PARTNER trial does not allow [us] to assess the performance of trans-apical TAVI in inoperable patients.
- In patients with severe aortic stenosis who are no [sic] candidates for surgery, TAVI significantly reduces the rate of death from any cause (ARR 20% at 1 year) as compared with standard therapy.
- In the Continued Access population (n = 90), TAVI had an absolute 12.7% higher mortality at 1 year as compared with standard therapy.
- Standard therapy included a balloon aortic valvuloplasty in most patients, a procedure considered as a palliative measure that has never been shown to be more effective than medical treatment.
- Stroke rate at 1 year was twice as high in TAVI patients compared to standard therapy (10.6% vs. 4.5%).
- In Cohort B, patients with prohibitive anatomical conditions were unevenly represented in both study groups. Subgroup analysis of those patients showed a more favourable effect of TAVI at 30 days (4.4% absolute survival difference) and after 1 year (8.8% absolute difference) compared to patients with medical prohibitive conditions.”
Internal Validity
- “Critical analysis of the methodology used in the PARTNER study indicates a rather high risk of bias, mainly in Cohort B.
- The unequal distribution of the basic characteristics between the study groups, to the advantage of TAVI, raises questions as to whether patient randomisation proceeded correctly. The randomisation procedure is only described in brief in the study protocol and our requests for further explanation from the study sponsor did not provide additional clarity. The fact that the main author of the study had significant financial interests in demonstrating the efficacy of TAVI raises eyebrows.
- Furthermore, the unexpected results of the Continued Access study that were conflicting with those of the pivotal trial raise questions.
- The so-called “standard therapy” involved an aortic balloon valvuloplasty in 84% of the patients in Cohort B. According to international practice guidelines, this form of treatment can sometimes be justified as an approach to treat aortic stenosis in the extreme elderly, but is anything but the standard. It is actually also a highly invasive technique with its own inherent severe risks. Its added value with respect to strictly medical treatments has never been demonstrated.
- In the elderly with severe aortic valve stenosis and severe co-morbidities, any procedure performed on the aortic valve should be considered as a palliative therapy. Such treatment decisions are determined by the question as to whether the quality of life of the patient in question, with his/her additional severe non-cardiac problems, can be expected to improve. This was not sufficiently demonstrated in the PARTNER study.”
Physicians’ Preferences
- “A physician’s performance in estimating the operative risk of a patient with aortic stenosis and significant co-morbidities has not been clearly established and may be subject to bias. In this respect, ethical questions come into play. Depending on the physicians’ preferences, less sick patients may be treated by TAVI although they could reasonably have open AVR. On the other hand, some patients may be offered TAVI although their co-morbidities preclude any significant improvement in their quality of life with a correction of the aortic stenosis. In a recent comment, the FDA deplores that whereas the PARTNER trial protocol defined patients who should not have surgery due to extensive co-morbidities, it did not actively consider patients who should not have TAVI.”
Transcatheter Aortic Valve Implantation for Aortic Stenosis (NICE July 25, 2011)
National Institute for Health and Clinical Excellence (NICE). Interventional procedures programme. Interventional procedure overview of transcatheter aortic valve implantation for aortic stenosis. 25 July 2011. Online at: http://www.nice.org.uk/nicemedia/live/11914/55669/55669.pdf
The National Institute for Health and Clinical Excellence (NICE) reviewed its 2008 guidance and provided an overview of transcatheter aortic valve implantation for aortic stenosis (April 2011). NICE sought additional consultation comments (July 2011) and is reassessing the procedure prior to issuing new interventional procedure guidance which is currently pending publication.
At the time of the April 2011 overview, NICE noted that in the U.K., TAVI is usually performed in patients who are very ill and are therefore inappropriate for conventional surgery, so TAVI is usually palliative in intent. Long-term data are lacking, with maximum median follow-up of 3.7 years. NICE also noted that “it is difficult to compare outcomes in non-randomized comparative studies since patients who are selected for TAVI are likely to be more ill and more likely to suffer complications or die.” Additionally, “Advisers stated that the procedure should only be performed by interventional cardiologists or cardiac surgeons with outstanding interventional experience and skills. The importance of a multidisciplinary team of surgeons and cardiologists when performing was highlighted.”
Transcatheter Aortic Valve Implantation for Aortic Stenosis: Guidance (NICE March 28, 2012)
National Institute for Health and Clinical Excellence (NICE). Interventional procedures programme. IPG421 Transcatheter aortic valve implantation for aortic stenosis: guidance. 28 March 2012.
This document replaced the Interventional Procedure Guidance (IPG) 266 on transcatheter aortic valve implantation for aortic stenosis and included the following points:
1.1 “Evidence on the safety of transcatheter aortic valve implantation (TAVI) for aortic stenosis shows the potential for serious but well-recognised complications.”
1.2 “For patients with aortic stenosis who are considered to be unsuitable for surgical aortic valve replacement (SAVR; see sections 1.6 and 2.1.3) the evidence on the efficacy of TAVI is adequate. For these patients, TAVI may be used with normal arrangements for clinical governance, consent and audit. Details of all patients should be entered into the UK Central Cardiac Audit Database.”
1.3 “For patients with aortic stenosis for whom SAVR is considered suitable but to pose a high risk (see sections 1.5, 1.6 and 2.1.3) the evidence on the efficacy of TAVI is inadequate. For these patients TAVI should only be used with special arrangements for clinical governance, consent and data collection or research. NICE encourages clinicians to enter suitable patients into the UK TAVI trial. In addition, details of all patients should be entered into the UK Central Cardiac Audit Database.”
1.4 “For patients with aortic stenosis for whom SAVR is considered suitable and not to pose a high risk (see sections 1.6 and 2.1.3) the evidence on the efficacy of TAVI is inadequate. For these patients TAVI should only be used in the context of research. NICE encourages clinicians to enter suitable patients into the UK TAVI trial. In addition, details of all patients should be entered into the UK Central Cardiac Audit Database.”
1.5 “Clinicians wishing to undertake TAVI for patients with aortic stenosis for whom SAVR is considered suitable but to pose a high risk (see section 1.3) should take the following actions. Inform the clinical governance leads in their Trusts. Ensure that patients understand the risk of stroke and death, and the uncertainty about the procedure's efficacy in the long term. Provide them with clear written information. In addition, the use of NICE's information for patients ('Understanding NICE guidance') is recommended.”
1.6 “Patient selection should be carried out by a multidisciplinary team including interventional cardiologists, cardiac surgeons, a cardiac anaesthetist and an expert in cardiac imaging. The multidisciplinary team should determine the risk level for each patient.”
1.7 “TAVI is a technically challenging procedure that should be performed only by clinicians and teams with special training and experience in complex endovascular cardiac interventions. Units undertaking this procedure should have both cardiac and vascular surgical support for emergency treatment of complications.”
2.1.3 “Patients may be unsuitable for SAVR because of medical comorbidities or because of technical considerations (for example, if the patient has a calcified aorta or scarring from previous cardiac surgery) which mean that the risks of SAVR outweigh the potential benefits. Patients who are suitable for SAVR range from those considered to be high risk (for example, as defined in the PARTNER A trial) to those for whom the benefits of surgery clearly outweigh the risks of surgery.”
2.5.3 “The Committee recognised that many patients with severe aortic stenosis have a poor prognosis as a result of comorbidities. It regarded careful overall assessment of life expectancy as an important
consideration when selecting patients for TAVI.”
2.5.4 “The Committee noted that a range of different devices are available for this procedure and there may be differences in clinical outcomes following the use of these different devices, for example, the need for subsequent pacemaker insertion.”
California Technology Assessment Forum (2012)
Tice J. Health technology assessment. Transcatheter aortic valve replacement in patients with severe aortic stenosis who cannot undergo surgery: a technology assessment. California Technology Assessment Forum. 8 February 2012. Online at: http://www.ctaf.org/content/assessment/detail/1426
In its technology assessment, the California Technology Assessment Forum (CTAF) website noted that five criteria were used “to determine if a medical technology improves health outcomes and is safe and effective:”
- The technology must have final approval from the appropriate government regulatory bodies.
- The scientific evidence must permit conclusions concerning the effectiveness of the technology regarding health outcomes.
- The technology must improve the net health outcomes.
- The technology must be as beneficial as any established alternatives.
- The improvement must be attainable outside the investigational settings.
For criterion two, evidence is graded using the following criteria:
Level 1: Randomized trials that had enough power to demonstrate a statistically significant health outcome;
Level 2: Randomized trials with results that were not statistically significant but where a larger trial might have shown a clinically important difference;
Level 3: Nonrandomized concurrent cohort comparisons between contemporaneous patients;
Level 4: Nonrandomized historical cohort comparisons between current patients and former patients (from the same institution or from the literature);
Level 5: Case series without control subjects.
CTAF searched the Medline, Embase, Cochrane databases and the Database of Abstracts of Reviews of Effects from 1945 to December 2011. Case series were included in the TA if at least 100 patients were treated with TAVR. Nineteen articles were identified including 13 case series, two comparative trials and one randomized trial (no mention was made of the remaining three articles). The TA reviewed evidence from studies using either the CoreValve or the Sapien valve or both. In addition, evidence was reviewed regardless of the route of administration: transfemoral, transapical, transaortic or subclavian. The author noted, however, that “[D]ata for the SAPIEN valve deployed using the transfemoral approach is most relevant for this assessment because the current FDA indication is for this delivery approach.”
The author determined that all five TA criteria were met. For criterion two, the levels of evidence were graded as levels one, three and five; further discussion regarding these assignments was not provided in the TA. The author concluded that the “case-series data and the small comparative studies gave inadequate information to fully understand the relative benefits and harms of TAVR compared with standard therapy.”
Regarding the one identified randomized trial (PARTNER B), the author stated that the “trial was not methodologically perfect. Neither the patients nor the outcome assessment was blinded. There were baseline differences between the two groups indicating that the TAVR group had a lower overall risk and fewer important comorbidities, such as COPD. Concerns have been raised because 64% of patients in the standard therapy group received aortic valvuloplasty within 30 days of randomization and an additional 20% after 30 days. However, none of these issues are of sufficient magnitude to explain the large one-year mortality difference between the two groups.
Patient selection is essential to ensure that the results of the PARTNER trial apply to patients treated in the community. All patients must be eligible for the transfemoral approach. A multidisciplinary team that includes at a minimum one cardiac surgeon, a general cardiologist, and an interventional cardiologist should agree that a patient is inoperable before offering TAVR. Patients must be informed of the upfront risk of death, stroke, pacemaker placement, and major vascular complications (16% in the PARTNER B trial). Patients also need to be informed that the long-term durability of the percutaneous aortic valve remains unclear. Observational data from one study suggest that patients who survive the first year following TAVR do well during the following year, but more data are needed. There is a high prevalence of moderate to severe AR, which may lead to recurrent symptoms or unforeseen problems with the valve. As was highlighted by Dr. Lazar in his editorial on the PARTNER B trial, given these uncertainties TAVR should not be performed in patients with a long life expectancy until more data are available. Additional studies are also needed before extending the use of TAVR to other patient groups and to other delivery approaches. Finally, as was highlighted at the February 2012 CTAF meeting, the dispersion of this technology to new centers across the United States must proceed with careful thought given to training and proctoring multidisciplinary teams to become new centers of excellence. Attention needs to be paid to appropriate patient selection, their pre-operative evaluation, surgical techniques, and post-operative care in order to preserve and improve upon the results attained in the PARTNER B trial. As described under TA 5, the specialty societies are collaborating to ensure that this happens in a rational and comprehensive manner.”
The TA recommended the use of the Sapien transcatheter aortic valve “for the treatment of severe, symptomatic aortic stenosis in patients determined to be inoperable by a cardiac surgeon who can be treated using the transfemoral approach.”
3. Internal Technology Assessment
CMS searched PubMed from 2000-2012 for randomized controlled trials (RCTs), substudies of such RCTs regarding QoL; technology assessments, systematic reviews and clinical guidelines which featured or included the pivotal PARTNER trial; device complications; plus prospective registries of consecutively enrolled patients reporting long-term outcomes, with keywords including symptomatic, severe aortic stenosis, transcatheter, aortic valve, implantation and/or replacement. We additionally searched for recent studies and reviews of frailty. To define the standard therapy to which TAVR has been compared in published clinical trials, we include recent reviews of open surgical and minimally invasive aortic valve replacement in the Medicare population. Studies must have been published in peer-reviewed English language journals. Abstracts, voluntary registries, and studies with less than 50 patients (unless reporting a significant adverse event not published elsewhere) were excluded. The literature search was limited to the English language and specific to the human population, but included studies conducted in all countries, including the United States. Public access information from the FDA website was also used.
Evidence Summary
Standard Open and Minimally Invasive Surgical AVR
Melby S, Zierer A, Kaiser S, et al. Aortic valve replacement in octogenarians: risk factors for early and late mortality. Annals of Thoracic Surgery 2007 May;83(5):1651-1656; discussion 1656-1657.
Melby and colleagues from the Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis concluded: “Patients aged 80 years and older who undergo AVR have acceptable short-term and long-term survival regardless of NYHA status. Concomitant CABG [coronary artery bypass grafting] improved operative and long-term survival in this population. Despite their increased age, aggressive surgical treatment is warranted for most patients.”
Filsoufi F, Rahmanian P, Castillo J, et al. Excellent early and late outcomes of aortic valve replacement in people aged 80 and older. Journal of the American Geriatrics Society 2008 February;56(2):255-261.
Filsoufi and colleagues from Mount Sinai School of Medicine in New York concluded: “Excellent results after AVR can be expected in patients aged 80 and older, with minimal increase in postoperative mortality and acceptable postoperative morbidity. Respiratory failure is the main postoperative complication in patients aged 80 and older. Recent advances in operative techniques and perioperative management have contributed to better surgical outcomes in these patients than found in historical reports.”
Thourani V, Myung R, Kilgo P, et al. Long-term outcomes after isolated aortic valve replacement in octogenarians: a modern perspective. Annals of Thoracic Surgery 2008 November;86(5):1458-1464; discussion 1464-1465.
Thourani and colleagues from the Emory University School of Medicine in Atlanta concluded: “In the modern era, octogenarians have acceptable short- and long-term results after open AVR. Comparisons of less invasive techniques for AVR should rely on outcomes based in the modern era and decisions regarding surgical intervention in patients requiring AVR should not be based on age alone.”
ElBardissi A, Shekar P, Couper G, et al. Minimally invasive aortic valve replacement in octogenarian, high-risk, transcatheter aortic valve implantation candidates. Journal of Thoracic and Cardiovascular Surgery 2011 February;141(2):328-335.
ElBardissi and colleagues from the Brigham and Women's Hospital and the Harvard Medical School in Boston concluded: “Patients thought to be high-risk candidates for surgical aortic valve replacement have excellent outcomes after minimally invasive surgery with long-term survival that is no different than that of an age- and gender-matched U.S. population. These data provide a benchmark against which outcomes of transcatheter aortic valve implantation could be compared.”
Randomized Controlled Trials of Transcatheter AVR
Leon M, Smith C, Mack M, et al. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. New England Journal of Medicine 2010 October 21;363(17):1597-1607.
Smith C, Leon M, Mack M, et al. Transcatheter versus surgical aortic-valve replacement in high-risk patients. New England Journal of Medicine 2011 June 9;364(23):2187-2198.
The PARTNER study incorporated two parallel prospective, unblinded, randomized, active-treatment controlled, multi-center pivotal trials evaluating the safety and effectiveness of transcatheter aortic valve replacement, via transfemoral or transapical (Cohort A only) delivery, in a stratified population of high risk (Cohort A) or inoperable (Cohort B) patients.
Because the study enrolled two distinct populations, two cohorts were separately-powered and analyzed. As depicted in Figure 1, an initial stratification based on operability for AVR surgery was used to assign patients to either Cohort A or B. Assignment to cohorts was followed by determination of vascular access for transfemoral delivery. Patients who were considered high surgical risk and eligible for transfemoral access were stratified into Cohort A and randomized to treatment (transfemoral AVR) or control (surgical AVR). Cohort A patients who were not eligible for transfemoral access were evaluated as candidates for transapical delivery and, if appropriate, randomized to treatment (transapical AVR) or control (surgical AVR). Those patients who were considered non-surgical candidates were stratified into Cohort B and randomized to treatment (transfemoral AVR) or control (“standard” therapy). Those assigned to Cohort B who did not meet the criteria for transfemoral delivery were not enrolled in the study because the sponsor declined to have a transapical arm in Cohort B (PARTNER trial protocol 2009; FDA Executive Summary 2011).
Figure 1. PARTNER Trial Enrollment Diagram (FDA Executive Summary 2011)
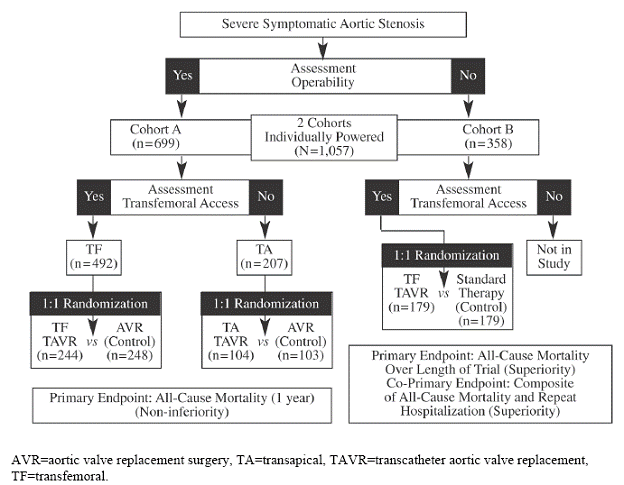
“The “standard” therapy control group predominantly consisted of subjects receiving BAV (78.2%); other patients received medical therapy alone (7.9%), AVR (6.1%), apical-aortic conduits (3.3%), or TAVR outside of the U.S. (2.2%)” (FDA Executive Summary 2011).
Candidates for the pivotal PARTNER trial were highly selected (1057 of 3105 (34%) patients who were screened at all the study centers underwent randomization) and each candidate must have met all of the following inclusion/exclusion criteria:
PARTNER Cohort A Inclusion Criteria
“All candidates for Cohort A of this study must meet all of the following Inclusion criteria:
- Patients must have co-morbidities such that the surgeon and cardiologist Co-PIs concur that the predicted risk of operative mortality is ≥ 15% and/or a minimum STS score of 10. A candidate who does not meet the STS score criteria of ≥ 10 can be included in the study if a peer review by at least two surgeon investigators (not including the enrolling surgeon) concludes and documents that the patient’s predicted risk of operative mortality is ≥ 15%. The surgeon's assessment of operative comorbidities not captured by the STS score must be documented in the study case report form as well as in the patient medical record.
- Patient has senile degenerative aortic valve stenosis with echocardiographically derived criteria: mean gradient > 40 mmHg or jet velocity greater than 4.0 m/s or an initial aortic valve area (AVA) of < 0.8 cm2 (indexed EOA < 0.5 cm2/m2). (Qualifying AVA baseline measurement must be within 45 days prior to randomization).
- Patient is symptomatic from his/her aortic valve stenosis, as demonstrated by NYHA Functional Class II or greater.
- The subject or the subject’s legal representative has been informed of the nature of the study, agrees to its provisions and has provided written informed consent as approved by the Institutional Review Board (IRB) of the respective clinical site.
- The subject and the treating physician agree that the subject will return for all required post-procedure follow-up visits.”
PARTNER Cohort B Inclusion Criteria
“All candidates for Cohort B of this study must meet # 2, 3, 4, 5 of the above criteria, and
- The subject, after formal consults by a cardiologist and two cardiovascular surgeons agree that medical factors preclude operation, based on a conclusion that the probability of death or serious, irreversible morbidity exceeds the probability of meaningful improvement. Specifically, the probability of death or serious, irreversible morbidity should exceed 50%. The surgeons' consult notes shall specify the medical or anatomic factors leading to that conclusion and include a printout of the calculation of the STS score to additionally identify the risks in these patients.”
PARTNER (Cohort A and B) Exclusion Criteria
“Candidates will be excluded from the study if any of the following conditions are present:
- Evidence of an acute myocardial infarction ≤ 1 month before the intended treatment (defined as: Q wave MI, or non-Q wave MI with total CK elevation of CK-MB ≥ twice normal in the presence of MB elevation and/or troponin level elevation (WHO definition).
- Aortic valve is a congenital unicuspid or congenital bicuspid valve, or is non-calcified.
- Mixed aortic valve disease (aortic stenosis and aortic regurgitation with predominant aortic regurgitation >3+).
- Any therapeutic invasive cardiac procedure performed within 30 days of the index procedure, (or 6 months if the procedure was a drug eluting coronary stent implantation).
- Pre-existing prosthetic heart valve in any position, prosthetic ring, severe mitral annular calcification (MAC), severe (greater than 3+) mitral insufficiency, or Gorelin syndrome.
- Blood dyscrasias as defined: leukopenia (WBC < 3000 mm3), acute anemia (Hb < 9 mg%), thrombocytopenia (platelet count < 50,000 cells/mm³), history of bleeding diathesis or coagulopathy.
- Untreated clinically significant coronary artery disease requiring revascularization.
- Hemodynamic instability requiring inotropic support or mechanical heart assistance.
- Need for emergency surgery for any reason.
- Hypertrophic cardiomyopathy with or without obstruction (HOCM).
- Severe ventricular dysfunction with LVEF < 20.
- Echocardiographic evidence of intracardiac mass, thrombus or vegetation.
- Active peptic ulcer or upper GI bleeding within the prior 3 months.
- A known hypersensitivity or contraindication to aspirin, heparin, ticlopidine (Ticlid), or clopidogrel (Plavix), or sensitivity to contrast media, which cannot be adequately premedicated.
- Native aortic annulus size < 18mm or > 25mm as measured by echocardiogram.
- Patient has been offered surgery but has refused surgery.
- Recent (within 6 months) cerebrovascular accident (CVA) or a transient ischemic attack (TIA).
- Renal insufficiency (creatinine > 3.0) and/or end stage renal disease requiring chronic
dialysis.
- Life expectancy < 12 months due to non-cardiac co-morbid conditions.
- Significant aortic disease, including abdominal aortic or thoracic aneurysm defined as maximal luminal diameter 5 cm or greater; marked tortuosity (hyperacute bend), aortic arch atheroma (especially if thick [> 5 mm], protruding or ulcerated) or narrowing (especially with calcification and surface irregularities) of the abdominal or thoracic aorta, severe “unfolding” and tortuosity of the thoracic
aorta (applicable for transfemoral patients only).
- Iliofemoral vessel characteristics that would preclude safe placement of 22F or 24F introducer sheath such as severe obstructive calcification, severe tortuosity or vessels size less than 7 mm in diameter (applicable for transfemoral patients only).
- Currently participating in an investigational drug or another device study. [Note: Trials requiring extended follow-up for products that were investigational, but have since become commercially available, are not considered investigational trials].
- Active bacterial endocarditis or other active infections.
- Bulky calcified aortic valve leaflets in close proximity to coronary ostia.”
( http://www.nejm.org/doi/suppl/10.1056/NEJMoa1008232/suppl_file/nejmoa1008232_protocol.pdf)
“Changes in the protocol were made after this unblinded study started enrollment, the most significant of which was the addition of a co-primary composite endpoint of mortality and hospitalization. The 6-minute walk test endpoint was also added after the start of the trial. The protocol was fully approved in March 2009 (Version 3.2) coincident with completion of enrollment into the Cohort B study, and approval to begin a Continued Access study. At the onset, the Cohort B continued access study protocol was the same as the randomized PARTNER study until Cohort A enrollment was completed. In August 2009, enrollment into the Cohort A study was completed, and the Continued Access study was expanded to allow enrollment of Cohort A subjects in a non-randomized protocol. Randomization for the Cohort B group was also discontinued at that time” (FDA Executive Summary 2011).
Figure 2. Timeline of PARTNER trial and the Continued Access study (Neyt 2011)
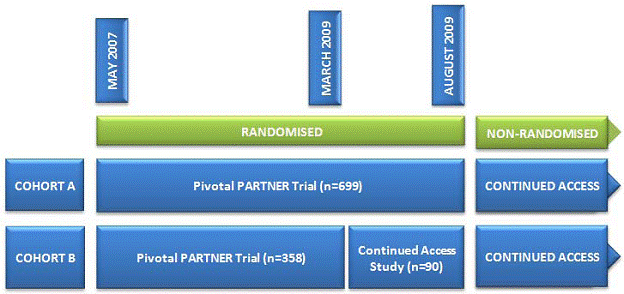
“Enrollment in the nonrandomised Continued Access cohort is ongoing. As of November 1, 2010, 160 nonrandomized patients have been enrolled” (Belgian HTA 2011).
PARTNER Cohort A
Smith C, Leon M, Mack M, et al. Transcatheter versus surgical aortic-valve replacement in high-risk patients. New England Journal of Medicine 2011 June 9;364(23):2187-2198.
At 25 centers in the United States (22 centers), Canada (2 centers), and Germany (1 center), 699 high-risk patients (348 TAVI versus 351 surgical aortic valve replacement) with severe aortic stenosis and cardiac symptoms (NYHA class II function or worse) – who were considered to be candidates for surgery despite the fact that they were at high surgical risk – were assigned to either transcatheter aortic valve replacement with a balloon-expandable bovine pericardial valve (either transfemoral or transapical approach) or surgical aortic valve replacement in an industry sponsored trial. As outlined in Cohort A’s inclusion criteria, “severe aortic stenosis was defined as an aortic-valve area less than 0.8 cm2 plus either a mean gradient of at least 40mm Hg or a peak velocity of at least 4.0 m per second.” High risk for operative complications or death determination was made by at least two surgeons at each center, using as a guideline a score of ≥ 10% on the STS risk model or due to the presence of coexisting conditions associated with ≥ 15% predicted risk of death by 30 days after surgery.
Mean age was 83.6 years for the TAVI group and 84.5 years for the surgical group; and females comprised 42.2% in the TAVI group and 43.3% in the surgical control group. Baseline characteristics were comparable between groups. Extensive exclusion criteria for both Cohort A and Cohort B of the PARTNER trial included: bicuspid or non-calcified aortic valve, coronary artery disease requiring revascularization, left ventricular ejection fraction (LVEF) <20%, aortic annulus < 18 mm or > 25 mm, severe mitral or aortic regurgitation, recent neurological event, and severe renal insufficiency. Patients in the transcatheter group underwent either transfemoral or transapical placement of the aortic valve, on the basis of whether peripheral arteries could accommodate the sheath. Randomization was achieved with the use of computer-generated randomized blocks at each site and for each subgroup. The primary end point was death from any cause at one year in the intention-to-treat population. The primary hypothesis was that transcatheter replacement is not inferior to surgical replacement. All patients were followed for one year, starting during the index hospitalization, 30 days, six months, one year, and then yearly. Crossover between the two groups was permitted only when findings during the assigned procedure suggested the alternate treatment. Pre-specified secondary end points included:
- Death from cardiovascular causes;
- NYHA functional class;
- Repeat hospitalization because of valve- or procedure-related clinical deterioration;
- Myocardial infarction;
- Stroke;
- Acute kidney injury;
- Vascular complications;
- Bleeding;
- 6-minute walk distance;
- Valve performance (assessed with echocardiography).
“In a retrospective analysis of neurologic events adjudicated by the clinical events committee, major stroke was defined by a score of at least 2 on the modified Rankin scale (which ranges from 0 to 6, with higher scores indicating greater disability).” A priori, the investigators determined sample size between the two groups, with the design to demonstrate non-inferiority. A sample of 650 patients would provide a power of at least 85% to show non-inferiority of the primary end point, assuming a 1-year death rate of 29% in the transcatheter group and 32% in the surgical group. Non-inferiority would be established if the upper limit of the one-sided 95% confidence interval for the between-group difference in mortality was less than 7.5 percentage points, with alpha of 0.05. Other sample size computations were done, such as considering transfemoral placement independently. Fisher’s exact test was used for categorical variables and continuous variables were compared with the use of Student’s t-test. Time-to-event analyses, based on available data, were done with the use of Kaplan-Meier estimates and compared between groups with the use of the log-rank test. A test for interactions was performed. All analyses were intention-to-treat (not as treated).
Four patients died during the procedure (three in the experimental group and one in the control). Sixteen patients in the TAVI group (4.6%) received conventional surgical repair. One patient in the surgical group underwent transapical replacement. Multiple transcatheter valves (≥ 2) were implanted in seven patients due to difficulties, three of these patients died. Among seven other patients with similar difficulties, transcatheter placement was aborted in two patients, and was converted to open surgery in five patients. Late interventions in the TAVI group included another procedure (valvuloplasty) for paravalvular regurgitation in two patients and conversion to transapical placement in one patient. Additionally, patients in the TAVI group received heparin during the procedures and aspirin and clopidogrel for six months after the procedure.
Serious events were adjudicated by an independent committee, and a complete account of the “Clinical Outcomes at 30 Days and 1 Year in the Intention-to-Treat Population” for patients in Cohort A (Table 2 from Smith 2011) can be found in Appendix B at the end of this decision memorandum. Additional outcomes included:
Patient functional measures
- At 30 days, more patients in the experimental group had a reduction in symptoms to NYHA class II or lower.
- At 30 days, for patients who were able to perform the 6-minute walk test, patients in the transcatheter group walked farther than those in the surgical group (NICE 2011).
- At one year, the earlier between-group differences were not evident.
Echocardiographic data
- Aortic-valve gradients and area improved significantly after the two procedures at both 30 days and one year, as expected.
- At one year, transcatheter replacement was slightly superior to surgical replacement with respect to the mean aortic-valve gradient and mean valve area.
- Moderate or severe paravalvular regurgitation was more frequent in the transcatheter group than in the surgical group at 30 days (12.2% vs. 0.9%) and at one year (6.8% vs. 1.9%) (P < 0.001 for both
comparisons).
The authors concluded: “In high-risk patients with severe aortic stenosis, transcatheter and surgical procedures for aortic-valve replacement were associated with similar rates of survival at 1 year, although there were important differences in periprocedural risks.”
Kodali S, Williams M, Smith C, et al. Two-year outcomes after transcatheter or surgical aortic-valve replacement. New England Journal of Medicine 2012 March 26. [Epub ahead of print]
In Cohort A of the PARTNER trial (Smith 2011), 699 high-risk patients with severe aortic stenosis were randomly assigned to undergo either TAVR (N=348) or surgical AVR (N=351); and all patients have been followed for at least two years (median, 727 days; maximum, 1490 days). At two years, there was no significant difference between the TAVR and surgical AVR groups in either all-cause mortality (33.9% and 35%, respectively; P = 0.78) or cardiovascular mortality (21.4% and 20.5%, respectively; P = 0.80); and there was no significant difference in rate of repeat hospitalization between TAVR and surgical AVR (24.7% and 21.7%, respectively; P = 0.41). Between one and two years, there were 32 additional deaths with TAVR and 25 with surgical AVR. Between one and two years, there were four additional strokes with TAVR and four with surgical AVR, as well as two additional TIAs with TAVR and one with surgical AVR. At two years (Table 1), the frequency of all neurologic events (strokes and TIAs) thus remained higher with TAVR than with surgical AVR (11.2% and 6.5%, respectively, P = 0.05).
Moderate or severe paravalvular aortic regurgitation was more common after TAVR than after surgical AVR (7.0% versus 1.9% at one year, and 6.9% versus 0.9% at two years; P<0.001 for both comparisons). From the total of 348 patients who were randomized to TAVR, among the 143 patients who underwent echocardiographic evaluation two years after TAVR, paravalvular aortic regurgitation was unchanged in 46.2%, was improved in 31.5% and was worse in 22.4% of this limited number of patients with echocardiographic data both post-procedure and at two years. Presence of paravalvular or total aortic regurgitation (mild, moderate or severe versus none or trace) after TAVR was associated with increased late mortality (hazard ratio, 2.11; 95% CI, 1.43 to 3.10; P<0.001); and while the effect of regurgitation on mortality was proportional to the severity of regurgitation, even mild aortic regurgitation was associated with an increased rate of late deaths.
In conclusion, the frequency of all neurologic events (strokes and TIAs) at two years was significantly higher following TAVR than with surgical AVR (P = 0.05); paravalvular aortic regurgitation was more frequent after TAVR than with surgical AVR (P<0.001); and even mild paravalvular aortic regurgitation was associated with increased late mortality (P<0.001).
PARTNER Cohort B
Leon M, Smith C, Mack M, et al. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. New England Journal of Medicine 2010 October 21;363(17):1597-1607.
From 25 participating centers in the United States (21 centers), Canada (3 centers), and Germany (1 center), 358 high-risk patients (179 TAVI versus 179 control patients) with severe aortic stenosis and cardiac symptoms – who were not considered to be suitable for surgery – were enrolled at 21 sites (17 in the United States) and were randomly assigned to either transcatheter aortic valve replacement (AVR) with a balloon-expandable bovine pericardial valve (either a transfemoral or a transapical approach) or standard therapy (including balloon aortic valvuloplasty but not conventional surgery) in an industry sponsored trial. As outlined in Cohort B’s inclusion criteria, severe aortic stenosis was defined as an aortic-valve area less than 0.8 cm2 and either a mean gradient of at least 40mm Hg or a peak velocity of at least 4.0m per second. These patients were considered not to be suitable candidates for surgery due to the presence of coexisting conditions that could be associated with a predicted probability of 50% or more of either death by 30 days after surgery or a serious irreversible condition, as agreed upon by at least two surgeons.
Mean age was 83.1 years for the TAVI group and 83.2 years for the control group, and females comprised 54.1% in the TAVI group and 54.1% in the standard therapy control group. Baseline characteristics were not balanced and included several between-group differences that were statistically significant (p < 0.05):
- Lower logistic EuroSCORE in the TAVI group (p = 0.04);
- More patients with COPD in the control group (p = 0.04);
- More patients with atrial fibrillation in the control group (p = 0.04); and
- More patients with an extensively calcified aorta in the TAVI group (p = 0.05).
Exclusion criteria included: bicuspid or non-calcified aortic valve, acute myocardial infarction, substantial coronary artery disease requiring revascularization, LVEF < 20%, aortic annulus < 18mm or > 25 mm, severe mitral or aortic regurgitation, recent neurological event, and severe renal insufficiency. Patients in the transcatheter group underwent transfemoral placement of the valve. Randomization was achieved with the use of computer-generated randomized blocks at each site and for each subgroup. The primary end point was death from any cause at 1 year in the intention-to-treat population. “The co-primary end point was the rate of a hierarchical composite of the time to death from any cause or the time to the first occurrence of repeat hospitalization (after the index procedure) due to valve-related or procedure-related clinical deterioration.” The primary hypothesis was that transcatheter replacement is superior to standard therapy. All patients were followed for one year, starting during index hospitalization, 30 days, six months, one year, and then yearly. Crossover was not permitted. Pre-specified secondary end points included:
- Death from cardiovascular causes;
- NYHA functional class;
- Repeat hospitalization because of valve- or procedure-related clinical deterioration;
- Myocardial infarction;
- Stroke;
- Acute kidney injury;
- Vascular complications;
- Bleeding;
- 6-minute walk distance;
- Valve performance (assessed with echocardiography).
A priori, the investigators determined sample size between the two groups, with the design to demonstrate superiority. A sample of 350 patients would provide a power of at least 85% to show superiority of the primary end point, assuming a one year death rate of 37.5% in the transcatheter group and 25% in the control group. The analysis of the co-primary endpoint was performed using a nonparametric method. The sample size of 350 patients with a power of 95% was estimated on the basis of the co-primary endpoint. Fisher’s exact test was used for categorical variables and continuous variables were compared with the use of Student’s t-test. Time-to-event analyses, based on available data, were done with the use of Kaplan-Meier estimates and compared between groups with the use of the log-rank test. A two-sided alpha level of 0.05 was used for all superiority testing. All analyses were intention-to-treat (not as treated).
Two patients randomized to TAVI died before receiving the intervention. Four patients in the TAVI group did not receive a valve due to technical difficulties. Despite being categorized as unsuitable for surgery, twelve patients underwent AVR (conventional surgery), another five underwent two procedures (placement of a conduit from the left ventricular apex to the descending aorta plus AVR), and four underwent TAVI at a non-participating site outside the United States. Three patients in the TAVI group underwent repeat TAVI to treat clinically significant aortic regurgitation. All patients in the TAVI group received heparin during the procedures, and aspirin and clopidogrel for six months after the procedure.
Serious events were adjudicated by an independent committee, and a full account of the “Clinical Outcomes at 30 Days and 1 Year” for patients in Cohort B (Table 2 from Leon 2010) can be found in Appendix B at the end of this decision memorandum. Additional outcomes included:
Patient functional measures:
- At one year, 74.8% of surviving patients with TAVI as compared to 42.0% of the surviving patients with standard therapy had a reduction in symptoms to NYHA class II or lower.
- At one year, of the subgroup of patients able to perform the 6-minute walk, an analysis showed that there was significant improvement after TAVI and no change after standard therapy.
Echocardiographic data:
- At one year, the improvement in aortic-valve area and mean valve gradient was maintained.
- Moderate or severe paravalvular regurgitation was present in 11.8% of the patients in the TAVI group at 30 days and in 10.5% at one year. The incidence of moderate or severe transvalvular aortic regurgitation was 1.3% at 30 days and 4.2% at one year among patients in the TAVI group, as compared to 16.9% and 15.2% in the control group where the procedure for some was balloon valvuloplasty.
The authors concluded: “In patients with severe aortic stenosis who were not suitable candidates for surgery, TAVI, as compared with standard therapy, significantly reduced the rates of death from any cause, the composite end point of death from any cause or repeat hospitalization, and cardiac symptoms, despite the higher incidence of major strokes and major vascular events.”
Makkar R, Fontana G, Jilaihawi H, et al. Transcatheter aortic-valve replacement for inoperable severe aortic stenosis. New England Journal of Medicine 2012 March 26. [Epub ahead of print]
In Cohort B of the PARTNER trial (Leon 2010), 358 patients underwent randomization to either transfemoral TAVR (N = 179) or to standard therapy (N=179) which often included balloon aortic valvuloplasty. At baseline, there were significantly more patients with an extensively calcified aorta (P = 0.05) in the TAVR group, more patients with COPD (P = 0.04) and atrial fibrillation (P = 0.04) in the standard therapy group, and more than twice the number of patients (28 versus 12 patients) in the TAVR group as compared to standard therapy with an STS score < 5%. At two years, all-cause mortality was 43.3% following TAVR and 68.0% after standard therapy (P < 0.001); cardiac death rates were respectively 31.0% and 62.4% (P < 0.001); and TAVR’s survival advantage at one year remained significant in those surviving beyond the first year (hazard ratio, 0.58; 95% confidence interval [CI], 0.36 to 0.92; P = 0.02).
Overall, the rate of stroke was higher after TAVR than after standard therapy both at one year (11.2% versus 5.5%, P = 0.06) and at two years (13.8 versus 5.5%, P = 0.01) including, in the first 30 days, more ischemic events following TAVR (6.7% versus 1.7%, P = 0.02) and thereafter more hemorrhagic strokes with TAVR than with standard therapy (2.2% versus 0.6%, P = 0.16). At two years, the rate of rehospitalization was 35.0% following TAVR and 72.5% after standard-therapy (P < 0.001); and TAVR was associated with improved functional status as compared with standard therapy (P < 0.001). From a total of 179 patients who were randomized to transfemoral TAVR, among the 61 patients in the TAVR group for whom results of baseline, year one, and year two echocardiographic studies were available, paravalvular aortic regurgitation improved in 42.6%, did not change in 41.0%, and worsened in 16.4%. Stratification by STS scores ( < 5%, 5 to 14.9%, and ≥ 15%) was significantly associated with two-year mortality, i.e., the survival benefit of TAVR diminished with higher STS scores (P = 0.01).
In PARTNER Cohort B, the baseline characteristics of the TAVR and standard therapy groups were imbalanced. There was a wide range of STS scores demonstrating that technical or anatomical reasons for inoperability (extensively calcified or porcelain aorta, radiation, and chest wall deformity) were most common in the STS < 5% category; and patients with low STS risk scores ( < 5%), deemed inoperable largely due to such technical or anatomical factors, exhibited the most pronounced mortality reduction with TAVR (Makkar 2012, Figure 2 and supplementary appendix Figure 6). Conversely, higher STS scores and the presence of extensive coexisting medical conditions attenuated TAVR’s survival benefit. The authors concluded that the ultimate value of TAVR for symptomatic patients with aortic stenosis will depend in part upon “the careful selection of patients who are not candidates for surgery and who do not have extensive coexisting conditions that might overwhelm the benefits of TAVR and render the intervention futile.”
Quality of Life
Reynolds M, Magnuson E, Lei Y, et al. Health-related quality of life after transcatheter aortic valve replacement in inoperable patients with severe aortic stenosis. Circulation 2011 November 1;124(18):1964-1972.
Reynolds and colleagues performed a prospective health-related quality of life (HRQoL) substudy among patients enrolled in the PARTNER trial. In this publication they presented only the results of the PARTNER trial’s Cohort B – those patients who were not considered candidates for surgical valve replacement and who were therefore randomized to either TAVR or the standard therapy control group. In the PARTNER trial, HRQoL was assessed at baseline and then at one, six, and 12 months with the Kansas City Cardiomyopathy Questionnaire (KCCQ) and the 12-item Short Form 12 General Health Survey (SF-12). The KCCQ has undergone validation in heart failure patients, and its summary score has a range of 1-100, with higher being a better score. At baseline between treatment groups, the KCCQ overall summary (including symptoms, physical limitation, social limitation, and quality of life) was not different statistically, nor was either of the two components of the SF-12 (physical and mental). Baseline scores for the KCCQ and SF-12 were low and subsequently improved in both groups, though the improvement was greater in the TAVR group as compared to controls at all measured time points, with both clinical and statistical difference. The authors concluded: “Among inoperable patients with severe aortic stenosis, compared with standard care, TAVR resulted in significant improvements in health-related quality of life that were maintained for at least 1 year.”
Observational Studies: Long-Term Outcomes
Moat M, Ludman P, Belder M, et al. Long-term outcomes after transcatheter aortic valve implantation in high-risk patients with severe aortic stenosis. The U.K. TAVI (United Kingdom Transcatheter Aortic Valve Implantation) registry. Journal of the American College of Cardiology 2011 November 8;58(20):2130-2138.
The United Kingdom (U.K.) established an all-inclusive transcatheter aortic valve implantation registry to report outcomes of all TAVI procedures performed within that country. A total of 25 centers throughout England and Wales developed active TAVI programs between January 2007 and December 2009, and data were collected prospectively on 870 patients undergoing 877 TAVI procedures up until December 31, 2009. Two technologies were available to these units: the Medtronic CoreValve system and the Edwards SAPIEN valve. Since there are few data on outcomes beyond one year, Moat and colleagues’ (2011) publication was an attempt to address outcomes to date. Mortality tracking was achieved in 100% of patients with survival status reported as of December 12, 2010; and follow-up ranged from 11 months to 46 months.
The authors reported that “ Survival at 30 days was 92.9%, and it was 78.6% and 73.7% at 1 year and 2 years, respectively. There was a marked attrition in survival between 30 days and 1 year. In a univariate model, survival was significantly adversely affected by renal dysfunction, the presence of coronary artery disease, and a nontransfemoral approach; whereas left ventricular function (ejection fraction < 30%), the presence of moderate/severe aortic regurgitation, and chronic obstructive pulmonary disease remained the only independent predictors of mortality in the multivariate model.”
In the discussion section, Moat and colleagues further described that the “high attrition in the first year post-implant is also seen in the SOURCE registry and the Italian registries and in both cohorts of the PARTNER trial; for example, 18% of patients died after a TAVI between 30 days and 1 year in PARTNER A. It is of interest that there was an almost identical rate of attrition in the control (AVR) group.” The authors additionally noted that the incidence of early stroke was comparable to other registries and to the PARTNER trial, as well as that “the finding of magnetic resonance imaging evidence of (albeit seemingly silent) cerebral perfusion defects in 84% of TAVI patients highlights the need to evaluate neurological outcomes in these patients, including cognitive function. Embolic protection devices may have a role in ameliorating the incidence of stroke, but at present it remains a major concern and represents an obstacle to the application of TAVI in lower risk patients.”
Moreover, “in 61% of patients, there was a degree of paravalvular AR [aortic regurgitation] that would traditionally have been regarded as suboptimal or even unacceptable after AVR. The finding that the degree of post-implant AR was an independent predictor of survival at 1 year is an important observation and requires further detailed study. Whether the regurgitation is responsible for this adverse outcome or is merely a marker for other adverse features cannot be assessed from this registry. The presence of moderate or severe AR was more common in the Medtronic CoreValve cohort. There is some evidence that the degree of AR remains stable or even reduces during the first year post-implant. The influence of this residual AR on parameters such as the incidence of endocarditis and hemolysis and the effect on LV mass regression are unknown and will need to be further evaluated. A reduction in the incidence and severity of paravalvular AR represents an obvious target for technical improvements in the design of transcatheter valves and of implantation techniques.”
The authors also acknowledged that “the observation that COPD was an independent predictor of outcome is perhaps surprising. In patients with aortic stenosis and COPD, it can be difficult to be certain as to the precise contribution of each pathology in an individual patient with progressive severe breathlessness. For patients in whom COPD predominates [COPD was significantly greater in controls compared to TAVI patients in the PARTNER trial’s Cohort B], the relief of aortic stenosis may not change the clinical outcome as much as in other patient groups, and that may in part explain this observation.”
In conclusion, Moat and colleagues stated that “Midterm to long-term survival after TAVI was encouraging in this high-risk patient population, although a substantial proportion of patients died within the first year.”
Complications
Abdel-Wahab M, Zahn R, Horack M, et al. Aortic regurgitation after transcatheter aortic valve implantation: incidence and early outcome. Results from the German transcatheter aortic valve interventions registry. Heart 2011 June;97(11):899-906. PMID: 1398694
Abdel-Wahab and colleagues concluded that “significant AR [aortic regurgitation] after TAVI is common and is associated with increased in-hospital mortality. Long-term follow-up is critical to further define the impact of residual AR on clinical outcome. Until these data become available, every effort should be made to prevent and treat this complication.”
Frailty
Makary M, Segev D, Pronovost P, et al. Frailty as a predictor of surgical outcomes in older patients. Journal of the American College of Surgeons 2010 June;210(6):901-908.
Makary and colleagues from the John Hopkins University School of Medicine in Baltimore concluded: “Frailty independently predicts postoperative complications, length of stay, and discharge to a skilled or assisted-living facility in older surgical patients and enhances conventional risk models. Assessing frailty using a standardized definition can help patients and physicians make more informed decisions.”
Afilalo J. Frailty in patients with cardiovascular disease: why, when, and how to measure. Current Cardiovascular Risk Reports2011 October;5(5):467-472.
Afilalo from McGill University in Montreal concluded: “Frailty and CVD [cardiovascular disease] share common biological pathways, and CVD may accelerate the development of frailty. Frailty is identified in 25% to 50% of patients with CVD, depending on the frailty scale used and the population studied. Frail patients with CVD, especially those undergoing invasive procedures or suffering from coronary artery disease and heart failure, are more likely to suffer adverse outcomes compared to their non-frail counterparts. The 5-m gait speed test is a simple and effective way of objectively measuring frailty in patients with CVD and should be incorporated in risk assessment. Further research will clarify how to best incorporate frailty in existing risk models and how to optimize health status and prevent adverse outcomes in frail patients.”
Zenilman M, Chow W, Ko C, et al. New developments in geriatric surgery. Current Problems in Surgery 2011 October;48(10):670-754.
Zenilman and colleagues from Johns Hopkins Medicine in Baltimore reported that: “Frailty as a marker of a patient’s ability to tolerate stress has been validated by its ability to predict complications following surgery – the greatest stress test a person can withstand”, as well as that “The prevalence of frailty among those over age 65 has been estimated as between 7% and 16% and is more common among women and African American individuals. Among those presenting for elective surgery over age 65, the prevalence has been estimated at 11% being frail, and 41% being at least intermediately frail.”
4. Medicare Evidence Development & Coverage Advisory Committee (MEDCAC)
CMS did not hold a MEDCAC meeting on this topic.
5. Evidence-Based Clinical Guidelines
No evidence-based clinical guidelines for TAVR are presently available.
6. Professional Society Position Statements
ACCF/AATS/SCAI/STS Expert Consensus Document on Transcatheter Aortic Valve Replacement (2012)
Holmes D, Mack M, Kaul S, et al. 2012
ACCF/AATS/SCAI/STS Expert Consensus Document on Transcatheter Aortic Valve Replacement. Journal of the American College of Cardiology 2012;59:XX-XX. 59(13): 1200-1254 PMID: 22300974 Available online: http://content.onlinejacc.org/cgi/reprint/j.jacc.2012.01.001v1.pdf
This document was developed as an Expert Consensus Document (ECD) by the American College of Cardiology Foundation (ACCF), American Association for Thoracic Surgery (AATS), Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons in collaboration with the American Heart Association (AHA), American Society of Echocardiography, European Association for Cardio-Thoracic Surgery, Heart Failure Society of America, Society of Cardiovascular Computed Tomography, Society of Cardiac Magnetic Resonance, Society of Cardiovascular Anesthesiologists, and Mended Hearts.
ECDs are intended to inform practitioners, payers, and other interested parties of the opinion of ACCF and cosponsors concerning evolving areas of clinical practice and/or technologies that may be new to the practice community. Topics discussed by ECDs are so designed because the evidence base, experience with technology, and/or clinical practice are not sufficiently well developed to be evaluated by the formal ACCF/AHA Practice Guidelines process.
In summary, the potential risks and benefits of alternative treatment recommendations need to be carefully evaluated and discussed with the patient and family; and a team-based approach to patient care is a “foundational requirement” of TAVR. “Given the high-risk profile of patients, who often have multiple comorbidities, as well as the technical complexity of the procedure involved, this team-based care will need to include multiple contributors at different stages in the process but will be mainly centered around the primary cardiologist, the cardiovascular surgeon, and the interventional cardiologist. Patients and families must be included in the care team. Other team members will include cardiac anesthesiologists, heart failure specialists, structural heart disease physicians, imaging specialists and the nursing care team, among others.”
Tommaso C, Bolman R, Feldman T, et al. SCAI/AATS/ACCF/STS multisociety expert consensus statement: operator & institutional requirements for transcatheter valve repair and replacement; part 1 TAVR. Journal of the American College of Cardiology 2012 February 29 [Epub ahead of print]. PMID: 22387052
On January 10, 2012, the ACC submitted a letter to CMS accompanied by a preliminary guidance on Institutional and Operator Requirements for TAVR supported by multiple specialty medical societies – ACC, AATS, SCAI and the STS. The letter outlining the preliminary guidance, available in Appendix C of the proposed decision memorandum, has since been replaced by updated guidance available in a manuscript accepted for publication in the Journal of the American College of Cardiology.
The authors stress the importance of collaboration between the interventional cardiologists and cardiac surgeons in establishing a TAVR program. While they further note that, “the correlation between operator experience and performance metrics for these procedures has yet to be established,” they do offer a set of prerequisite skills and ultimately, volume requirements. In addition to the operators, there is an institutional commitment required by the hospital to ensure an infrastructure with the needed diagnostic imaging and therapeutic facilities. This includes a cardiac catheterization laboratory or hybrid operating room with a fixed radiographic imaging system, echocardiographic lab, vascular lab and computer tomography lab, post-procedure intensive care with personnel experienced in managed open-heart valve patients and overall, the hospital must provide support (including financial) to the team and program. While the document includes an extensive discussion of the above, Table 1 identifies the important requirements for a successful TAVR program:
| TABLE 1 |
TRANSCATHETER AORTIC VALVE REPLACEMENT |
| INSTITUTIONAL INTERVENTIONAL PROGRAM |
1000 CATH/400 PCI PER YEAR* |
| TAVR INTERVENTIONALIST |
100 STRUCTURAL PROCEDURES LIFETIME OR 30 LEFT SIDED STRUCTURAL PER YEAR OF WHICH 60% SHOULD BE BALLOON AORTIC VALVULOPLASTY (LEFT SIDED PROCEDURES INCLUDE EVAR, TEVAR, BALLOON AORTIC VALVE (BAV), AORTIC VALVE (AV) AND MITRAL VALVE (MV) PROSTHETIC LEAK CLOSURES AND VSD CLOSURES. (ATRIAL SEPTAL DEFECT/PATENT FORMAN OVALE (ASD/PFO) CLOSURE ARE NOT CONSIDERED LEFT SIDED PROCEDURES) SUITABLE TRAINING ON DEVICES TO BE USED |
| INSTITUTIONAL SURGICAL PROGRAM |
50 TOTAL AVR PER YEAR OF WHICH AT LEAST 10 AORTIC VALVE REPLACEMENT (AVR) SHOULD BE HIGH-RISK (STS SCORE ≥6) MINIMUM OF 2 INSTITUTIONALLY-BASED CARDIAC SURGEONS IN PROGRAM (MORE THAN 50% TIME AT HOSPITAL WITH SURGICAL PROGRAM) |
| TAVR SURGEON |
100 AVR CAREER, AT LEAST 10 OF WHICH ARE “HIGH-RISK” (STS SCORE ≥ 6) OR 25 AVR PER YEAR OR 50 AVR IN 2 YEARS AND AT LEAST 20 AVR IN LAST YEAR PRIOR TO TAVR INITIATION EXPERIENCE WITH, AND MANAGEMENT OF PERIPHERAL CARDIOPULMONARY BYPASS EXPERIENCE WITH OPEN RETROPERITONEAL EXPOSURE OF, AND SURGICAL INTERVENTION ON THE ILIAC ARTERIES ALL CASES MUST BE SUBMITTED TO A SINGLE NATIONAL DATABASE |
| EXISTING PROGRAMS |
30 TAVR (TOTAL EXPERIENCE) ONGOING CONTINUING MEDICAL EDUCATION (CME) (OR NURSING/TECHNOLOGIST EQUIVALENT) OF 10 HOURS PER YEAR OF RELEVANT MATERIAL ALL CASES MUST BE SUBMITTED TO A SINGLE NATIONAL DATABASE |
| TRAINING |
CARDIOLOGISTS MUST BE BOARD CERTIFIED/ELIGIBLE IN INTERVENTIONAL CARDIOLOGY SURGEONS MUST BE BOARD CERTIFIED/ELIGIBLE IN THORACIC SURGERY ADDITIONAL OPERATORS WHO ARE TRAINED OR EXPERIENCED IN STRUCTURAL HEART DISEASE AND HAVE UNRESTRICTED HOSPITAL PRIVILEGES IN STRUCTURAL PROCEDURES, MAY ALSO BE PART OF THE INTERVENTIONAL OPERATING TEAM WITH THE INTERVENTIONAL CARDIOLOGIST AND CARDIOVASCULAR SURGEON |
| NEW PROGRAMS |
20 TAVR PROCEDUES/YEAR or 40 TAVR PROCEDURES OVER 2 YEARS 30-DAY ALL-CAUSE MORTALITY <15% 30-DAY ALL-CAUSE NEUROLOGIC EVENTS INCLUDING TRANSIENT ISCHEMIC ATTACK (TIA’S) <15% MAJOR VASCULAR COMPLICATION <15%** >90% INSTITUTIONAL FOLLOW-UP
60% 1-YEAR SURVIVAL RATE FOR NON-OPERABLE PATIENTS (COHORT B) – AFTER THE PROGRAM HAS BEEN RUNNING FOR 2 YEARS (2-YEAR AVERAGE) ONGOING CME (OR NURSING/TECHNOLOGIST EQUIVALENT) OF 10 HOURS PER YEAR OF RELEVANT MATERIAL ALL CASES MUST BE SUBMITTED TO A SINGLE NATIONAL DATABASE |
| TRAINING |
1) CARDIOLOGISTS MUST BE BOARD CERTIFIED/ELIGIBLE IN INTERVENTIONAL CARDIOLOGY 2) SURGEONS MUST BE BOARD CERTIFIED/ELIGIBLE IN THORACIC SURGERY 3) ADDITIONAL OPERATORS WHO ARE TRAINED OR EXPERIENCED IN STRUCTURAL HEART DISEASE AND HAVE UNRESTRICTED HOSPITAL PRIVILEGES IN STRUCTURAL PROCEDURES, MAY ALSO BE PART OF THE INTERVENTIONAL OPERATING TEAM WITH THE INTERVENTIONAL CARDIOLOGIST AND CARDIOVASCULAR SURGEON |
* With acceptable outcomes for conventional procedures compared to NCDR (National Cardiovascular Data Registry) benchmarks
** According to VARC-2 (Valve Academic Research Consortium) definitions
7. Public Comments
During the initial 30-day public comment period (09/28/2011 - 10/28/2011), CMS received 30 public comments. All comments were generally supportive of coverage for TAVR; however some commenters strongly suggested that coverage be left to local Medicare contractors and not be handled through the NCD process. One commenter supported coverage in clinical trials. Several commenters cited concerns regarding access due to aspects of the formal request surrounding center of excellence requirements, facility requirements, staffing requirements and volume thresholds. Several commenters offered suggestions on how the formal request could be revised to address some of these concerns.
CMS received seven comments from professional societies and education and advocacy associations; seven from individuals who did not identify an associated organization or profession; six from medical facilities, physicians and researchers; four from hospital administrators; three from lawmakers; and three from device manufacturers.
The comments can be viewed in their entirety on our website at https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=257&ExpandComments=n&ver=3&NcaName=Transcatheter+Aortic+Valve+Replacement+(TAVR)&bc=ACAAAAAAIAAA&.
Public Comment Period 02/02/2012 – 03/03/2012
During the 30-day comment period following the release of the proposed decision memorandum, CMS received 83 comments. All commenters support coverage of TAVR but differ on various aspects of the proposed decision. One commenter does not support utilizing the NCD process, instead favoring the local coverage determination (LCD) process.
California Technology Assessment Forum (CTAF)
We received several comments regarding the CTAF. The commenters noted that CMS did not include this assessment in the decision memorandum. Several commenters cited the CTAF assessment to note that on label use of TAVR meets CTAF criteria for safety and effectiveness and improvement in net health outcomes.
CMS Response: We reviewed the CTAF and included a summary of the final CTAF document in section VII of this decision memorandum. As the CTAF authors noted, patients need to be informed that the long term durability of percutaneous aortic valve remains unclear. In addition, there is a high prevalence of moderate to severe aortic regurgitation after TAVR. The authors state, and we agree, that additional studies are needed to answer outstanding questions about TAVR.
Comments on other published materials
In contextualizing the outcomes of PARTNER’s Continued Access Patients and discussing the survival benefit of TAVR more generally, a commenter suggested Figulla, et al.’s (2011) systematic review in 2010 [minus the PARTNER trial] to “provide objective evidence on the efficacy and safety of [TAVR] at one-year follow-up and to assess whether [TAVR] confers a survival benefit compared with medical therapy.”
CMS Response: CMS reviewed the systematic review by Figulla (2011). This systematic review was not cited as evidence by CMS because the review did not include the pivotal PARTNER trial. Given the size of the PARTNER trial, we believe that TAVR reviews that do not include it are significantly incomplete and unrepresentative of the current evidence base and thus are less persuasive in our review of TAVR.
One commenter cites a study (Hayashida K, et al. JACC 2012;59;566-571) of 260 TAVR patients, more than half of whom were women, to note that better survival was seen in women at one year. This commenter also notes that women had better baseline characteristics and it is important to allow these types of studies to continue under the final policy.
CMS Response: We agree and believe these types of studies are important; and this national coverage determination allows for studies enrolling women to continue to be covered.
Expert Consensus Document
One commenter notes that new evidence-based guidelines have been published and references the 2012 ACCF/AATS/SCAI/STS Expert Consensus Document on TAVR (Holmes 2012).
CMS Response: CMS discussion regarding expert consensus documents is included in the professional society statements section of this decision memorandum.
Indications
Three commenters support linking coverage of TAVR to FDA approved indications as this will allow coverage to evolve as TAVR technology and practice evolves without the need for repeated NCD reconsiderations. Another commenter supports linking coverage with the FDA approval process to support further study of a promising new technology.
CMS response: We agree and appreciate the support for this final decision.
A commenter contends that the evidence is more than adequate for CMS to conclude that TAVR is reasonable and necessary for appropriately selected beneficiaries, when provided by heart teams in appropriate facilities, with outcomes reported through a qualified registry.
CMS response: For reasons explained throughout this decision memorandum, we believe that TAVR is reasonable and necessary in the context of research under section 1862(a)(1)(E).
One commenter asserts that CMS should cover off-label uses in circumstances where the commenter claims there is patient benefit. Another commenter contends that coverage should not be restricted to FDA approved indications because he/she believes this will impair physicians’ ability to deliver appropriate care.
CMS response: CMS supports off-label uses of TAVR when certain protections are in place (see Section I). We believe it is important for CMS to support the evidence development when appropriate. The decision also supports broader coverage if newly developed evidence leads the FDA to expand the labeled the indications. We also believe that claims of “appropriate” care, if unsupported by adequate evidence, are speculative until evidence is actually developed. Moreover, patients may be at risk if the existing co-morbidities would preclude the expected benefit from correction of the aortic stenosis.
One commenter agrees that TAVR should be covered only for severe aortic stenosis, but asserts that requiring the other proposed conditions of coverage would be too restrictive.
CMS response: We disagree. The evidence that led to the approval of the aortic stenosis indication was generated within of restrictive context of a clinical trial and thus we believe is not generalizable to patients outside a clinical study. We believe that the conditions of coverage are reasonable and compatible with the FDA requirements of postapproval data collection, as well as the professional society requestors consensus statements, and are appropriate to enhance the likelihood that beneficiaries who undergo TAVR will have successful clinical outcomes.
One commenter requests that CMS revise language in section A(1) “for this indication” as that phrase suggests that CMS will not support coverage for any TAVR device and any use outside the labeled indications under any circumstances.
CMS response: CMS supports coverage of unlabeled indications in clinical studies.
One commenter requests coverage and prospective registry inclusion of high risk cohorts not specifically addressed in PARTNER, recommending that the PARTNER trial exclusion criteria not be used to identify coverage or non-coverage.
CMS response: The final decision does not prohibit enrollment of high risk cohorts meeting the labeled indications in the registry. We believe the exclusion criteria in PARTNER prevent us from generalizing PARTNER reported results to populations that would be ineligible to participate in PARTNER at this time. Should such cohorts fall under the FDA approved label in the future, they would be included under section A1 of the final decision.
One commenter requests CMS clarify the distinction between “on-indication” and “on-label” uses of TAVR, and one commenter requests that CMS clarify the difference between coverage for “FDA approved indications” and conditional “coverage for all unlabeled uses.”
CMS response: We revised the decision to use the term indication rather than the term label.
Many commenters referenced specific patient circumstances without specific reference to either indication or label. Four commenters contend that CMS should cover other approaches or access routes than transfemoral and one commenter requests that patients not be excluded from coverage due to the presence of an aortic aneurysm or the inability to gain femoral access. Two commenters request coverage for “valve-in-valve” patients and two commenters request coverage for ESRD patients under CED. One commenter requests coverage for patients with untreated clinically significant coronary artery disease requiring concomitant revascularization. One commenter contends that beneficiaries of advanced age be allowed freedom of choice in treatment regardless of STS score or co-morbidities. One commenter requests coverage for the use of CTA of the heart/chest, abdomen and pelvis for assessing patient suitability pre-TAVR and for procedural TAVR planning. One commenter notes that, for patients with severe LVH secondary to aortic stenosis, it is difficult to ensure the patients do not have hypertrophic cardiomyopathy. Another commenter suggests specifying that for patients with significant aortic disease that abdominal aortic or thoracic aneurysms are untreated and one commenter suggests adding “temporary iliac conduit” to the qualifying approaches under iliofemoral vessel characteristics. One commenter requests that CMS identify how long non-covered indications will extend.
CMS response: For certain rapidly evolving technologies, we agree that there are important advantages to linking closely with the FDA approval process. Under subsection A, coverage with registry participation is for patients satisfying the FDA approved indications. For example, the revised policy would no longer categorically noncover valve-in-valve patients or patients with hypertrophic cardiomyopathy. We also cover certain off –indication uses, subject to specific restrictions in a CMS approved trial.
We do not believe the current FDA indication encompasses the patient that, according to a surgeon’s evaluation, is otherwise an operable candidate but would rather select TAVR as the course of treatment, and is therefore outside the scope of this decision. As noted in its November 2011 press release “it is not approved for patients who can be treated with open-heart surgery.” We find no evidence or other justification to support an age-based exemption from evidence-based coverage and FDA approval conditions. While we have made changes in the final decision to require the hospital have as part of its infrastructure certain imaging abilities, we do not believe it is appropriate to designate a specific type of pre-operative imaging modality without evidence.
Coverage with Evidence Development
Outcomes
Two commenters contend that the KCCQ should be used for quality of life (QoL) measures and that requiring the collection of QoL data through five years is problematic due to missing data that may not be random. Commenters suggested that data should be collected at baseline, 30 days and one year follow-up for health status evaluations. One commenter requests that CMS provide specific direction on how QoL should be addressed in the registry.
CMS Response: CMS agrees that collecting data for 5 years may be problematic. There are challenges associated with collecting long term QoL data. However, we believe QoL information is crucial to the patient being able to make an informed decision regarding TAVR. Our final policy states that a qualifying registry will follow the patient for a minimum of one year rather than five. One year is when the patient generally has a follow up visit with his/her physician and therefore would be the most appropriate time to collect the QoL element. We have determined to not be more prescriptive regarding QoL due to the lack of better validated tools than the KCCQ.
One commenter suggests that national database operators be instructed to look broadly across the spectrum of QoL measures.
CMS Response: We appreciate the supportive comments.
One commenter asserts that stroke should be captured without differentiation between major and minor.
CMS Response: We agree that major and minor stroke can be combined simply as stroke.
Two commenters request that CMS specify that follow-up includes a combination of follow-up coupled with linked CMS MedPar data and the Social Security Master Death File to allow for thorough tracking. One commenter contends that the outcomes to be tracked require precise definition and suggests consideration of work done on TAVR by VARC (Valve Academic Research Consortium).
CMS Response: We agree it would be ideal to use Medicare claims data and the Social Security Master Death File. We expect that the data elements collected will allow linkage at the patient level with registry data. The matching of Medicare claims data and the Social Security Master Death File are not requirements of this final decision. CMS agrees that registries should include a data dictionary for interpretative purposes and believes that it is appropriate for defining outcomes, and we anticipate that a CMS approved national TAVR registry will have a standardized, well developed data dictionary.
One commenter asserts that if the final decision does not include outcome definitions, the specific outcomes be vetted through a stakeholder advisory group.
CMS Response: Medicare does have a mechanism for public vetting of clinical evidence through the Medicare Evidence Development & Coverage Advisory Committee (MEDCAC) and it is plausible that when enough registry information and analyses are available that such a committee meeting will be scheduled.
IDE Coverage
One commenter requests that CMS modify section B of the proposed decision in a manner that allows IDE trials to continue. Another commenter requests that CMS clarify that the CED policy is not intended to interfere with coverage of TAVR in IDE trials. One commenter urges CMS to maintain coverage for FDA approved Category B IDE trials.
CMS Response: In order to be covered by Medicare, a TAVR device that is designated as a category B device must meet all the CED requirements specified under this NCD. Medicare contractors are bound by this national coverage determination by our regulations at 42 CFR 405.211, and are not permitted to provide coverage in an IDE trial that does not meet these standards. The removal of the superiority requirement should facilitate coverage of Category B devices under this NCD.
One commenter asserts that IDE trials should be excluded from the superiority requirement and one commenter contends that IDE trials should be exempt from center and physician requirements.
CMS Response: As discussed more fully below, we have revised the decision to remove the superiority requirement and to permit non-inferiority study designs.
Superiority Requirement
Twenty three commenters disagree with the language in section B requiring that unlabeled uses of TAVR be covered in clinical studies that have superiority designs. Eight commenters request that CMS remove the superiority requirement from the final policy and the remainder of commenters offer various reasons for why the superiority requirement should be removed.
Commenters assert that the superiority requirement is unnecessarily restrictive and will inhibit the medical device industry from introducing next generation devices. They state that superiority designs will not result in meaningful studies and will slow the advance of TAVR in the US; the requirement contradicts past CMS and FDA policies in support of clinical research and may have negative public policy implications; Cohort A patients would be eliminated from participation in clinical studies, as would the ability for patients to choose their preferred treatment; the requirement will negatively impact cost, coverage, industry innovation, competition, and access to new therapies; all future trials will likely be non-inferiority and if only superiority trials are covered these trials will not be covered; the requirement sets a precedent that may negatively alter the course of future research and development; superiority designs may not include factors important to the congenital heart community; policies should not be used to discourage manufacturers from using non-inferiority designs as this could discourage innovation to improve products; such a requirement is inconsistent with past policies and guidance, may negatively impact trials currently underway, and may inappropriately deny beneficiary access to care.
One commenter contends that preemptively covering only superiority trials and not allowing coverage for non-inferiority trials is extremely limiting and will adversely impact development of future clinical evidence. One commenter requests that the Agency clarify if it plans to impose a superiority requirement in future NCDs and one commenter suggests that such a requirement may have something to do with a desire to avoid payment for IDE trials and subsidizing research that sponsors should perhaps be supporting, but notes that such a policy decision should not be made in this NCD.
CMS Response: While we believe superiority trial designs provide important advantages that are not completely addressed by non-inferiority design, we recognize that non-inferiority trials have a place in the conduct of medical device regulatory trials. Therefore, we believe a broad non-coverage of non-inferiority trials may have unintended consequences for certain important studies.
CMS originally proposed superiority in the interest of patient-centered care, because otherwise how do providers and patients get the information to know whether a treatment actually improves patient outcomes? All we know from a non-inferiority trial is whether a treatment is not worse than a comparator by more than some prespecified amount. For instance, in the latest extension to the Consolidated Standards of Reporting Trials (CONSORT) statement, Piaggio and colleagues (2006) emphasized that study design – be it superiority, non-inferiority or equivalence – should be appropriate to the question to be answered, and that trial reports must be clear enough to allow clinicians and patients to reliably interpret their results and conclusions.
Critically, non-inferiority trials aim to declare a new treatment acceptable, not to demonstrate actual improvement in clinical outcomes; and in such studies, patient cross-over (i.e., when patients switch from one study arm to another) may favor a new treatment, dampen differences versus the control treatment, and make it easier for trial sponsors to declare non-inferiority (Montovani 2010). Similarly, “the quality of trial conduct is critical to the validity of the inferences drawn in equivalence and non-inferiority trials. In any trial, it is quite possible for poor trial conduct to obscure differences between treatment groups. In a superiority trial, such problems would make the trial a failure. Thus in most cases there are very strong incentives to conduct a high-quality trial, to obtain high-quality data, and to have good compliance. But in equivalence and non-inferiority trials, these incentives are absent because the lack of difference is the desired outcome” (Siegel 2000).
As noted, by definition, a non-inferiority trial therefore attempts to demonstrate that a new treatment is not worse than a comparator by more than a pre-specified amount, i.e., the non-inferiority margin or delta; but “clinicians must know who has chosen the margin, and why” (Ricci 2010). Fleming and colleagues (2011) addressed Ricci’s question and, in addition to stating that “non-rigorous margins allow substantial risk for accepting inadequately effective experimental regimens, leading to the risk of erosion in quality of health care”, opined that:
“Commonly, there is a tension between wanting the non-inferiority margin, δ, to be large enough to allow for timely completion of the non-inferiority trial, and to be small enough to enhance trial integrity and the preservation of a substantial proportion of the demonstrated benefits provided by currently available regimens. To be candid, the choice of margins that are much too large often is not based on misunderstandings or differences of judgment between informed and unbiased clinicians and scientists, but rather on the clear recognition that wider margins allow sponsors to conduct smaller trials as well as trials that will have a substantially higher probability of providing ‘positive’ non-inferiority conclusions. Allowing margins to be chosen in such a manner is dangerous to public health interests since this allows substantial risk for erosion in the quality of health care through the replacement of effective standard regimens by experimental regimens having inferior benefit-to-risk profiles.”
Contrary to concerns about large sample size requirements for superiority study designs, CMS notes that Piaggio, et al.’s (2006) extension to the CONSORT statement found that – while sample sizes for non-inferiority and equivalence trials are “unfortunately” often too small – the required size of non-inferiority trials may actually be larger than for superiority trials. Likewise, in a cross-sectional study based on a random sample of 200 two-arm, parallel group superiority (100) and non-inferiority (100) RCTs published from 2004-2009 in 27 leading journals, non-inferiority trials required larger sample sizes, were more often conducted at multiple centers than superiority trials, and were more often industry funded (Gayet-Ageron 2010).
Additionally, one can test for non-inferiority or equivalence for primary outcomes at the same time as for superiority for secondary outcomes, e.g., harms, safety or convenience (Gotzsche 2006, Gayet-Ageron 2010). Nevertheless, while noting that non-inferiority testing allows for inclusion of superiority testing in the same study without need for adjustment of the statistical methods, Vavken (2011) cautioned regarding the potential weaknesses in non-inferiority testing:
“One is the potential to flood the healthcare market with ‘me too’ procedures and products that are non-inferior to current gold standard treatments but do not add additional value. Another potential pitfall is biocreep, i.e., the iterative process of establishing non-inferiority to the current gold standard of a slightly less effective, new treatment, followed by the use of this new treatment as a gold standard for an even newer, non-inferior but again slightly less effective treatment, and so on. Finally, methodologic rigor is even more important in non-inferiority than in superiority trials because of the problem of confusing non-inferiority with a Type I error. Briefly, this can be understood as follows: a superiority trial looks for a statistically different/clinically meaningful difference, and all flaws in study design and conduct make it harder to find such a difference, which makes this a conservative design (erring on the save [sic] side). Noninferiority looks for similarity, or ‘’no statistically different/clinically meaningful difference’’ as similarity cannot be tested directly. As in superiority trials, flaws in study design and conduct will make it harder to find a difference between treatments, but as‘’no difference’’ is the preferred outcome, noninferiority testing is anticonservative: a poorly designed and conducted noninferiority study has a greater chance of a false positive outcome, a Type I error.”
Finally, a study of non-inferiority trials published from 2005-2009 in the International Standard Randomized Controlled Trial Number (ISRCTN) or ClinicalTrials.gov trial registries reported that most registry records of non-inferiority trials do not mention non-inferiority design and do not include the non-inferiority margin (Dekkers 2011). To better inform patient centered care, clarity and interpretability of results is important. As such, to enable clinicians and patients to make better informed healthcare decisions about potential harms as well as benefits, CMS believes that:
- Where feasible, superiority study designs should be used to investigate non-approved, off-indication and off-label uses; and
- Where a non-inferiority or equivalence study design is utilized, trial sponsors should comply with the most recently published CONSORT checklist of items for reporting non-inferiority or equivalence trials, including specifying for each such trial on ClinicalTrials.gov prior to patient enrollment:
- That the trial is a non-inferiority or equivalence trial;
- The rationale for using a non-inferiority or equivalence design;
- For which outcomes non-inferiority (one-sided) or equivalence (two-sided) hypotheses apply, and for which superiority (two-sided) hypotheses apply;
- Both the trial’s prespecified delta [Δ or δ] and alpha [α]; and
- Whether a one- or two-sided confidence interval approach was used.
CED clarification and process
Ten commenters specifically address CED in their comments. One commenter contends that a clear implementation process for CED must be established using a steering committee with broad stakeholder representation and the final coverage decision should include an interim coverage policy, next steps for establishing a steering committee and a timeline for the committee to develop a data collection framework. Another commenter requests that CMS establish an open and transparent process involving stakeholders to implement appropriate registry management and oversight to ensure appropriate research questions are developed and the data collected is suitable to answer these questions.
CMS Response: The NCD process is an open and transparent process with specific statutory timeframes that allows the public to comment on the specific decision, including the CED elements of the decision. However, general comments regarding CED are outside the scope of this decision. We note, that CMS recently solicited public comments on CED and a MEDCAC is being held on this topic on May 16, 2012.
One commenter requests that the final policy in this decision include more specific information about the characteristics of studies likely to qualify for CED, and one commenter requests that CMS clarify the baseline to which clinical studies are expected to be compared.
CMS Response: Our CED requirement furnishes flexibility so different types of studies can be designed to address the questions outlined in the decision. Recognizing this, we believe that acceptable protocols may have unique characteristics, and we do not believe it would be appropriate to establish further prospective requirements at this time. Therefore, CMS believes it is appropriate to address each proposal individually as we have historically done for CED studies.
One commenter requests that CMS establish time limitations for CED.
CMS Response: We do not think CED time limits are appropriate at this time for this decision.
One commenter contends that CED, using prospective, registry-based, observational studies, should be used to extend coverage for off-label indications.
CMS Response: We disagree. We believe that broader coverage of off-label indications, i.e. indications that have not been approved by the FDA, are not currently supportable outside of formal clinical study protocols and that premature broad coverage could also undermine the FDA review process.
One commenter requests that coverage be limited to statistically robust, well-designed, appropriately sponsored trials because more evidence is necessary to address safety, sub-populations, durability and physician qualifications and expertise.
CMS Response: We agree.
A commenter requests that the requirement for existing TAVR centers to participate in RCTs or post-approval studies, A(3)(b) (i.e., experienced TAVR centers), be removed because it is inconsistent to impose such requirements on existing programs but not new programs.
CMS Response: Under the final decision, CED requirements are the same for hospitals with TAVR experience and hospitals without TAVR experience.
One commenter requests that CMS clarify whether A(3)(c)(i) (i.e., participation in a TAVR registry) means that the requirement is met by participation in the STS/ACC registry or ongoing research-based studies like PARTNER.
CMS Response: While the requirement can be fulfilled in a CMS approved registry, which may, if approved, include the STS-ACC registry, this provision does not preclude participation in a clinical study as a way to meet the CED requirement for an FDA approved indication. However, we do not require participation in a clinical study with the requirements specified under section B unless it is for uses that are not expressly listed as an FDA approved indication.
One commenter asserts that the requirement in B(1) to adhere to standards (a)-(m), as well as the subsequent language, be removed.
CMS Response: We disagree with removing the language specified in B3 as this language is essential to ensuring that studies are designed and conducted to answer questions relevant to Medicare beneficiaries and meet the relevant regulatory requirements. These criteria have consistently been included in proposed and final CED policies and have been vetted and agreed to by AHRQ. AHRQ support is required for any research under section 1142 of the Act, and as such, for Medicare coverage under CED when conducted pursuant to that authority.
CED Registry Requirement
One commenter asserts that all centers should participate in a prospective TAVR study and/or registry. Two commenters support the proposed registry reporting requirement, and one commenter contends that it is essential that the registry is tied to CED. One commenter supports close monitoring of TAVR outcomes through participation in a carefully monitored CED registry. One commenter supports the collection and review of individual patient outcome data in a registry format that is easily accessible and retrievable. One commenter supports the proposal that all TAVR patients be enrolled in a qualified prospective registry. One commenter supports the use of a prospective national outcomes database under CED as this is consistent with and builds upon recommendations from thought leaders during the FDA review and approval process.
CMS response: We appreciate the support for ongoing data collection.
One commenter requests that CMS provide specific information about what constitutes an “eligible” prospective national registry. One commenter requests clarification regarding how collected data will be utilized. One commenter asserts that the five year follow up should be removed. One commenter contends that CMS should be able to find TAVR reasonable and necessary and still maintain the registry requirement. One commenter requests removal of the registry requirement until a consensus on research questions complementing existing medical evidence with well defined endpoints as well as an agreement of study design have been reached, the amount of data collected is scaled to capture only data with any bearing on reasonable and necessary and the registry is made affordable, in the public domain and data is available to all stakeholders. One commenter asserts that the registry should be structured to ensure full stakeholder participation. One commenter supports the use of a registry but expresses concerns about associated costs and the amount of data included. One commenter contends that the registry fee is excessive and must be reevaluated if its use is mandated by CMS for reimbursement.
CMS Response: We believe the decision supports broad registry participation by stakeholders. In alignment with the FDA post-market study, we have changed the 5-year registry requirement to one year. We also understand the concerns set forth by commenters surrounding registry details. As we have noted elsewhere in this memorandum, we have determined that research on TAVR devices will be covered when furnished under specified conditions of coverage. We believe that there already exists a meaningful consensus on research questions and endpoints, and that this is reflected in the alignment of FDA postapproval study and CMS CED requirements, supported by the physician professional societies. Registry requirements are listed in section A 5 of the decision. CMS believes these requirements are the foundation to allow for the evidence questions to be answered and to qualify as CED for coverage. Pricing arrangements among external parties are beyond the scope of this memo.
One commenter contends that mandatory registry participation does not comply with requirements for obtaining informed consent for research under the HHS regulations for protection of human subjects at 45 CFR §46.116 as the proposed decision does not require informed consent to participate in the registry. This commenter also states that the informed consent for patient agreement to participate in this registry would be the result of coercion/undue influence and beneficiaries who refuse to consent to participate would be penalized by not receiving treatment. This commenter suggests revising A5 to include language asking patients to voluntarily enroll as well as language specific to the protection of human subjects regulation.
Two commenters ask CMS to confirm that it does not intend to require facilities to seek informed consent from patients for entry of data into the registry. These commenters note that the STS database receives no federal funds and is not engaged in federally regulated activities so it is not subject to the Common Rule and thus not required to obtain informed consent from parties. One commenter also notes that the ACC’s NCDR registries have qualified for a waiver of the informed consent requirement under 45 CFR Part 46. The two commenters further explain that the Office for Human Research Protections (OHRP) has clarified that sites collecting identifiable patient information in the course of clinical care that submit to external researchers are not engaged in human subjects research and thus not required to obtain informed consent to patients. Additionally the commenters note that OHRP has taken the position that where the Common Rule does apply to multi-center clinical trials or registries, central IRB review and approval of waiver requests is appropriate and local sites can rely on such approval. These commenters recommend changing language in A5 to require the registry operator to be responsible for ensuring compliance with all applicable regulations relating to the protection of human subjects (45 CFR Part 46 and 21 CFR Parts 50 & 56).
CMS Response: We appreciate the importance of human subjects protection as well as the opinions expressed by OHRP regarding under what circumstances informed consent is required or may be waived by an institutional review board. As such, we have inserted language into the final decision that addresses the protection of human subjects and the need for researchers and registry operators to ensure they are compliant with any regulations pertaining to the protection of human subjects. We have discussed with OHRP the issue of whether making participation in the registry a condition of coverage for TAVR would be a violation of the informed consent requirement under 45 CFR 46.116 that requires investigators to seek informed consent only under circumstances that minimize the possibility of coercion or undue influence. OHRP has clarified that such a condition of coverage would not violate the requirements of 45 CFR Part 46.
Quality of care and patient access issues
One commenter asserts that the NCD should mandate geographic-based patient based access that trumps criteria and guidelines in order to account for issues such as long travel distances, small hospitals and rural areas. One commenter suggests improving geographic access to TAVR by allowing multiple hospitals to collaborate in meeting institutional requirements. One commenter contends that institutional requirements will result in patients needing to travel significant distances for access to TAVR and create barriers to care for vulnerable populations. Two commenters contend that the final policy should consider patient access and safety. Another commenter notes that patient access may be limited due to health plan coverage limitations or an inability to pay out of pocket for travel expenses. One commenter contends that proposed requirements fail to recognize expertise and volume of specialized procedures performed in non-academic centers which would unrealistically require critically ill patients to travel to a limited number of regional centers.
CMS Response: We appreciate the challenges encountered by beneficiaries located in geographic areas with limited access to qualified medical institutions. We recognize that these challenges are not unique to TAVR, and that patients living in remote areas may travel significant distances to receive appropriate specialized care. However, we believe that the coverage criteria, including the facility and operator requirements incorporated in the final decision, are essential to ensure safety, maximize the benefit and minimize the risk of TAVR and appropriately balance geographic access issues. The relevant specialty societies have expressed a willingness to establish and maintain mentoring programs that will provide significant support to providers and practitioners that do not immediately meet the requirements of this NCD. We believe this approach is preferable to the alternative, which would otherwise have beneficiaries receive TAVR from providers and practitioners who have not yet demonstrated the ability to furnish the procedure and manage the related care successfully. We are hopeful that information from the registry may support future expansion of TAVR to geographically remote sites.
General Institutional and Operator Requirements
One commenter asserts that practitioner experience should be more important than site experience and one commenter notes that hospital and practitioner requirements should strike a balance between safe use of technology and patient access. One commenter recommends that the final policy involve a heart-team centric approach rather than focusing on individual operator and institution requirements. One commenter asserts that institutions and individuals already involved in TAVR should not be required to meet criteria and that they should automatically be approved to perform TAVR when the final NCD is released. One commenter asserts that institutional and individual surgeon and interventional cardiologist requirements should be applied to new TAVR programs and the performance measures from section A(3)(b)(iii) should be met after successful implementation of the program. One commenter requests that CMS clarify the center and physician credentialing process and allow centers to determine what physicians are appropriate for their TAVR programs.
One commenter contends that most of the proposed institutional and provider requirements are reasonable and one commenter asserts that volume and outcome requirements in section A(3)(b) should be removed. One commenter supports adoption of clinical competency criteria from professional society guidelines and one commenter supports safety protections for patients by restricting TAVR to the most capable practitioners and facilities. One commenter notes that specific training on TAVR should be differentiated from general education about TAVR and similar cardiac procedures which are not sufficient to meet training requirements. One commenter asserts that if CMS uses volume requirements a technical advisory panel should be formed including industry-wide stakeholders to analyze and define volume requirements and interim facility and provider requirements more consistent with the original TAVR NCA should be implemented in the mean time. One commenter contends that criteria for selecting qualified TAVR facilities and physicians should be reviewed and updated as experience is gained and technology advances and stakeholders should be involved in the process.
CMS Response: We have revised the proposed decision to focus the requirements more clearly on the heart team practitioners. We believe the final decision appropriately reflects the professional society consensus document as well as public comments to ensure beneficiaries receive safe and appropriate, high level care. Thus we do not believe that a technical advisory panel is needed at this time. We expect that professional societies, device manufacturers and thought leaders in this field will regularly evaluate their own requirements and standards set forth in the consensus document and keep CMS abreast of any changes in light of newly gained experience.
Five commenters expressed a belief that we require two cardiac surgeons deem a patient inoperable. One commenter requests that language be revised to indicate that the surgeons must determine operability or that the surgeons agree that the conditions are met to permit TAVR. One commenter contends that the requirement is unreasonable as many centers only have one cardiac surgeon on staff, and one commenter asserts that the requirement be consistent with FDA approval indications which require such a determination by one cardiac surgeon. One commenter requests that, if the two physician requirement remains, CMS specify that one may be an interventional cardiologist and that both physicians are not required to evaluate the patient in person. One commenter suggests that the requirement be changed to a panel of three medical doctors, including two surgeons and one cardiologist (not the referring cardiologist).
CMS Response: We did not propose that two surgeons deem a patient inoperable. We did propose that the patient be independently evaluated by two cardiac surgeons regarding suitability for surgical AVR. As the clinical evidence used for FDA approval and for much of this final decision was based on the PARTNER trial that required an evaluation and determination to be made by two cardiac surgeons, CMS believes this requirement is appropriate and consistent with available evidence.
Operator requirements
Thirty-seven commenters express disagreement with the interventionalist requirement of professional experience with 50 structural heart disease procedures. Four of these commenters request complete removal of the requirement while other commenters offer reasons why it is inappropriate and suggest alternative requirements. Fourteen of these commenters request that the number be reduced and/or various procedures be considered to meet this requirement including balloon aortic valvuloplasty (BAV), TAVR, peripheral vascular interventional procedures, ASD, VSD closures and other congenital heart procedures, percutaneous ventricular assist devices, therapeutic interventional cases, PCI and ICR management skill sets, vascular access management, use of AAA or TAA devices and PFO. Nine of the commenters contend that the proposed interventionalist requirement would reduce access to TAVR and six commenters assert that the requirements inappropriately limit the providers who can perform TAVR and the facilities in which TAVR may be performed.
Commenters also question the applicability of structural heart disease procedure experience to the skill set needed to perform TAVR. One commenter suggests focusing on the heart team approach rather than this requirement and one commenter asserts that patient outcome data relative to professional practice should be used.
Four commenters request that CMS clarify the definition of structural heart disease procedures; two commenters contend that the definition should include any structural heart disease; and one commenter requests that the final policy include examples of qualifying procedures.
One commenter contends that surgical AVR volume does not predict TAVR outcomes as demonstrated in the European experience and one commenter asserts that AVR requirements should be less burdensome with 50 career AVRs and 10 per year. One commenter contends that surgeons should have significant high risk (STS score > 10) surgical AVR experience of > 100 cases.
One commenter suggests that programs have board certified surgeons with extensive experience in aortic valve and root replacement, aortic dissection repair and bypass grafting. One commenter asserts that physician criteria are biased towards surgeons with less experience in catheter and wire based techniques. One commenter requests a clearer definition for institutionally based cardiac surgeon and that the definition should not be limited to physicians employed by hospitals. One commenter requests that CMS consider future TAVR providers in the NCD and one commenter suggests adding a provision for coverage for operators who are early in their career with limited experience but appropriate training. One commenter suggests that a track for well-trained and well-qualified young surgeons should be included in the final policy. One commenter asserts that objective requirements focusing on training and qualifications of physicians should be implemented.
One commenter suggests that interventional cardiologist requirements include experience performing diagnostic AS cases and larger experience performing coronary angioplasty. One commenter disagrees with the stringent cardiologist criteria and one commenter contends that the selection should be facility based. One commenter notes that interventional cardiologist criteria are not realistic and will limit expansion of TAVR to community based programs that otherwise meet proposed criteria. One commenter asserts that proposed interventional cardiologist requirements should be applied to the entire interventional cardiology TAVR heart team. One commenter recommends that interventional cardiologists should be performing an average of 75 PCI cases annually over the previous five years, and that the team may accept physicians with extensive structural heart disease procedural experience but no coronary experience. One commenter notes that interventional/structural cardiologists should be very experienced with BAV. One commenter requests that interventional cardiologists with significant structural experienced who are not certified in interventional cardiology (i.e. pediatric cardiologists) should be able to participate and have TAVR implantation privileges. One commenter requests that TAVR performed by BE/BC pediatric cardiologists with training and experience in structural heart disease interventions and who are part of the interventional operating team be included in coverage.
One commenter contends that volume is not the correct measure but that physicians should meet clinical outcome measures to focus on quality care prevent elimination of good teams from providing TAVR. Three commenters disagree with the use of individual volume criteria as many surgeons and interventionalists may not qualify to perform TAVR. One commenter contends that it would be meaningful to know the number of structural heart cases operators in the PARTNER and CoreValve trials had performed in order to estimate the number operators should have before becoming part of the heart team. One commenter requests the inclusion of a defined timeframe for surgeon and interventional cardiologist professional experience with procedures. One commenter supports operator volume and outcome standards and one commenter states that the amount of cases proposed is arbitrary. One commenter notes that the necessary skill set can be achieved by most interventional cardiologists and CT surgeons working together. One commenter requests clarification of the term “performed” in the context of physician qualification and expertise.
One commenter asserts that operators should perform at least 100 coronary interventional cases per year and one commenter contends that operators should have training, privileges, experience and skills in percutaneous treatment of peripheral arterial disease. One commenter asserts that interventional cardiologists and/or surgeons performing TAVR should be experienced with large bore arterial access and complication management. One commenter contends that extensive experience in aortic valvuloplasty is not appropriate given the current lack of indications. One commenter asserts that operators should be five years out in practice and have performed at least 1000 PCIs as primary operators with very low complication rates. One commenter contends that operators should have hospital privileges and at least five years of experience with peripheral vascular interventions. One commenter notes the absence of training requirements in the peripheral arteries which is the main area of complication for TAVR. One commenter asserts that operators should have performed 10 or more valvuloplasties before performing TAVR and one commenter notes that operators should have experience with complex coronary interventions. One commenter suggests that operators perform over 200 coronary and peripheral interventions per year.
CMS Response: While individual practitioners have expressed opinions contrary to the specialty societies, we find the consensus document generally more persuasive and representative of the specialty physician community. In response to the comments, we have made some revisions to the heart team volume criteria. Regarding interventionalists, we have included additional mechanisms to qualify with volume; left-sided procedures (which have also been better defined) or structural heart disease procedures. We have also specified the time frames for the experience. We have done the same with the cardiac surgeon requirements and have better described the required AVR experience. Changes have likewise been made to the hospital program requirements, increasing the catheterization volume while also changing the volume requirements for AVR and emphasizing that AVR volume must continue, though at a lesser degree, after a TAVR program is started. We believe the final policy, which focuses more closely on the heart team concept includes reasonable and appropriate requirements that will ensure patient safety and access while allowing qualified operators to participate on the team.
Institutional Requirements
One commenter contends that the 15 left-sided EVAR/TEVAR procedures per year should be defined as being performed by the team. One commenter asserts that the requirement should be for ≥ 15 left-sided structural interventions over two years and one commenter suggests at least 30 structural heart interventions should be required per year. One commenter contends that the EVAR/TEVAR requirement be delegated to the TAVR team and not just the interventional team and one commenter requests that EVAR and TEVAR performed by thoracic and cardiovascular surgery service count toward the ≥ 15 requirement.
Two commenters contend that the PCI requirements should be increased to 1000 per year. Three commenters assert that the requirements should be increased to 750-1000 per year, 300 per year and 500 per year, respectively. One commenter suggests increasing the cath/PCI requirement to 1000 per year. One commenter suggests increasing cath lab requirements to 400 diagnostic procedures and 150 interventional cases. One commenter recommends that minimum annual facility volume of 1000 cardiac catheterizations and 400 PCI cases be required. One commenter asserts that left heart structural procedures should replace PCI volume requirements. One commenter contends that there is no volume relationship in PCI and that PCI does not have major relevance to TAVR.
One commenter suggests that facility surgical AVR requirements be 75-100 per year. One commenter contends that 50 AVRs is arbitrary and no data indicates that facilities performing 25 per year would be less successful with TAVR. One commenter suggests that all valves be included in this requirement for a total of > 50 valve replacement procedures.
One commenter supports the volume requirements proposed in section A(3)(a) and A(3)(b); one commenter asserts that the institutional requirements should be met; and one commenter supports institutional volume and outcome standards. One commenter agrees with including volume and outcome standards from section A(3)(b)(iii) and suggests a review of the collected registry data. One commenter contends that the proposed requirements are biased toward university and training facilities and the exclusion of community hospitals will cause patients to suffer. One commenter asserts that the guidelines are overly restrictive and should be placed on teams not facilities as teams practice at multiple sites. One commenter notes that the volume requirements should be interpreted in the context of institution and team structure, character, function and outcomes. One commenter contends that clinical trial results should be monitored, but that outcomes should not be the sole determinate of a facility’s eligibility for coverage.
One commenter asserts that procedure volumes may correlate with outcomes but are not determinative of potential success of facilities performing TAVR. Two commenters note that proposed volume requirements are not supported by independent, evidence-based research; and one commenter contends that, as such, they are not a proper determination for facility qualification. One commenter recommends that CMS use parameters for facility equipment, monitoring, infrastructure requirements and other issues as in previous NCDs which would be more consistent with the recent TAVR NCA. One commenter requests further clarification and definition of all volume requirements.
One commenter suggests that institutions have intra-operative/intra-procedural echocardiography capabilities. One commenter recommends that programs should perform more than 350 open heart cases with at least three surgeons and also offer thoracic stent grafting and experience in vascular arterial repair. One commenter supports limiting coverage to institutions that perform ≥ 50 AVRs, ≥ 1000 catheterizations, and ≥ 20 PCIs (at least 20 of which are structural heart cases) in the year before initiation of a TAVR program. This commenter also contends that to maintain proficiency in cardiac care facilities should have minimum volumes of either ≥ 20 AVRs per year or ≥ 40 AVRs every two years. One commenter notes that experience with aortic stent grafting is a reasonable precursor to establishing TAVR programs but should not be an absolute pre-requisite. One commenter contends that new TAVR centers should demonstrate participation and reporting to a national TAVR registry, as well as patient follow-up, as part of their application. One commenter requests that CMS provide a more detailed explanation of how facility accreditation will be managed. One commenter requests more specificity on the length of required institutional patient follow-up and the type of information required.
CMS Response: We again appreciate the extensive feedback regarding specific aspects of particular volume and experience criteria in the proposed decision. As discussed in our response above, changes were made to the final decision based on the public comments received. In addition to changes reflected in the program volume requirements, we have included a narrative list to describe the facility infrastructure that is required of programs.
Heart Team
One commenter recommends placing more emphasis on the heart team concept and stressing site qualifications, team experience and team function. One commenter requests that the final policy better describe the heart team approach. One commenter contends that each facility should have documented establishment of the heart team consisting of cardiac surgeons; cardiologists with expertise in valvular heart disease, structural heart disease, interventional therapies, endovascular therapies, and cardiovascular imaging; and ad hoc inclusion of radiology, cardiac anesthesiology and other medical/surgical specialists. One commenter supports a team approach that assures each patient is thoroughly evaluated for care by a surgeon and interventional cardiologist. One commenter requests that language emphasizing that patients’ treatment preferences should be included in discussions regarding the decision to have surgical or interventional procedures and that treatment decisions should be individually based. One commenter asserts that guidelines should reinforce that the TAVR team demonstrate commitment to the heart team concept, receive training and have professional experience to complete TAVR procedures.
One commenter contends that the co-procedure requirement should be emphasized. One commenter asserts that a surgeon and a cardiologist on the heart team should both individually fulfill requirements in the published guidance document. One commenter recommends that the heart team be comprised of at least two engaged cardiothoracic surgeons and two interventional cardiologists at each site who lead and coordinate all clinicians to form a high-functioning multi-disciplinary heart team. This commenter suggests that support personnel including clinical cardiologists, echocardiographers, anesthesiologists, intensivists, valve clinic coordinators and other clinicians be made available as needed. This commenter notes that eligible heart teams should perform ≥ 25 AVRs and ≥ 75 PCIs in the year prior to initiating a TAVR program and maintain TAVR experience with at least ≥ 20 TAVRs a year or ≥ 40 every two years. This commenter also encourages CMS to adopt patient outcomes criteria for the heart team to be continually measured against in a national qualified TAVR registry.
CMS Response: We agree that the heart team is critical to ensuring TAVR is performed and provided appropriately and believe that the final coverage decision emphasizes the importance of the heart team and identifies requirements necessary for the team and its members. Most notably, we have allowed for the combined TAVR experience of the heart team to be sufficient to maintain necessary volumes (as opposed to creating separate volume criteria for the cardiac surgeon and interventionalist).
VIII. CMS Analysis
National coverage determinations (NCDs) are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1862(l) of the Act).
In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, section 1862(a)(1) of the Act in part states, with limited exceptions, no payment may be made under Part A or Part B for any expenses incurred for items or services:
- Which, are not reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member (§1862(a)(1)(A)) or
- in the case of research conducted pursuant to section 1142, which is not reasonable and necessary to carry out the purposes of that section. ((§1862(a)(1)(E)).
Section 1142 of the Act describes the authority of the AHRQ. Under section 1142, research may be conducted and supported on the outcomes, effectiveness, and appropriateness of health care services and procedures to identify the manner in which diseases, disorders, and other health conditions can be prevented, diagnosed, treated, and managed clinically.
Section 1862(a)(1)(E) of the Act allows Medicare to cover under coverage with evidence development (CED) certain items or services for which the evidence is not adequate to support coverage under section 1862(a)(1)(A) and where additional data gathered in the context of a clinical setting would further clarify the impact of these items and services on the health of Medicare beneficiaries. For your convenience, the 2006 CED guidance document is available at [http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads//ced.pdf].
As noted earlier, our review sought answers to the questions below. We have repeated them here for the convenience of the reader.
- Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for Medicare beneficiaries with severe symptomatic aortic stenosis who are not candidates for surgical aortic valve replacement? [PARTNER Cohort B]
- Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for high surgical risk Medicare beneficiaries with severe symptomatic aortic stenosis who are candidates for surgical aortic valve replacement? [PARTNER Cohort A]
If the answer to either or both of the questions above is positive, is the available evidence adequate to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from TAVR?
TAVR first-in-man was performed in 2002, and TAVR received Europe’s CE mark in 2007. Since then, it has been performed in patients at risk of complications from the surgical procedure though (considering the subjective nature of risk scoring) it is difficult to ascertain how many were “high-risk” as defined by the pivotal PARTNER trial. Prior to the PARTNER randomized trial, published evidence consisted of cases series and non-randomized comparative studies; and assessing mortality and morbidity in these studies is difficult due to patient selection bias, lack of standardized definitions for endpoints, variable center and operator experience, plus incomplete patient follow-up. “It is difficult to compare outcomes in non-randomised comparative studies since patients who are selected for TAVI are likely to be more ill and more likely to suffer complications or die” (NICE 2011).
The PARTNER Cohort A and Cohort B trials were randomized controlled trials designed for FDA pre-market approval and were designed to address certain indications, efficacy and patient safety issues. Of over 3,000 patients screened, 34% underwent randomization and were divided into two cohorts, all being very highly selected patients with severe symptomatic native aortic stenosis (AS). Partner A patients were those who were considered by at least two surgeons to be suitable for surgery (despite the fact that they were at high risk), whereas Cohort B patients were not considered to be suitable for surgery in the opinion of at least two surgeons. From that point, they were randomized (in Cohort A) to either surgical or transcatheter (either transfemoral or transapical) aortic valve replacement, and (in Cohort B) to either transcatheter (transfemoral) valve replacement or the standard therapy control group (78% of whom received balloon aortic valvuloplasty).
Viewing baseline characteristics as a reflection of randomization, while Cohort A was generally well matched, there were both anatomical and medical imbalances in Cohort B (extensively calcified aorta, COPD and atrial fibrillation). A superiority design was chosen for the inoperable Cohort B patients, with TAVR compared to standard therapy; and as noted above, most patients in the standard therapy group received balloon aortic valvuloplasty – a procedure which is not considered to be overly efficacious but rather a bridge to open or transcatheter treatment or palliation. However, for the operable high-risk Cohort A patients (Smith 2011), a non-inferiority study design was chosen. The concept of non-inferiority is not straightforward, and operationalizing this to patient care may be difficult. For instance, how does a provider accurately and adequately explain the risks and benefits of a new treatment to a patient which is reportedly “noninferior”? “The aim of a non-inferiority trial is to declare a new treatment acceptable, not to demonstrate improvement in clinical outcome.”(Mantovani 2010) That is, by definition, a non-inferiority trial attempts to demonstrate that a new treatment is not worse than a comparator by more than a pre-specified amount known as the non-inferiority margin or delta; but “clinicians must know who has chosen the margin, and why” (Ricci 2010). Patient-centered care therefore necessitates that adequate information be carefully and effectively communicated for informed decision making.
Cohort B
Though all patients in Cohort B were deemed by at least two surgeon investigators who had to agree that the patient was not a suitable candidate for surgery, twelve patients underwent conventional open AVR and had a rate of death at one year of only 33%. Historically, surgical risk was a decision based on the personal experience of the surgeon. There are a number of risk scoring systems that have been developed, but their usefulness is variable. Although many morbidity and mortality risk factors for these scores have been extensively analyzed, there continues to be uncertainty about who will experience adverse outcomes. This is especially true in the elderly (Saxton and Velanovich 2011). Attempts have been made to provide objective risk criteria, but considerable subjectivity of when and who should be operated on remains. Ultimately, any procedure is a risk-benefit decision, and it is important to accurately estimate this information for patient decision-making.
In Cohort B, the primary outcome was rate of death at one year from any cause; and TAVR did demonstrate superiority in the PMA trial. Also, the co-primary endpoint (a composite of death and recurrent hospitalization) was 42.5% with TAVI and 71.6% with standard therapy; but this composite endpoint was only added later and was not one of the trial’s original endpoints (FDA Executive Summary 2011). Among those who did survive to one year, the rate of cardiac symptoms (as judged by NYHA class) was lower among patients who had undergone TAVI than among those who received standard therapy. But optimal management of coronary artery disease (CAD) in the setting of TAVI has not been investigated, since patients with recent or untreated clinically significant CAD requiring revascularization (as well as significant peripheral vascular disease) were excluded from the PARTNER trial. It is altogether possible that patients who were excluded from the trial on the basis of comorbidities could benefit from TAVI.
One year mortality in the published PARTNER B trial was an absolute 20% lower for patients who underwent TAVI as compared to standard therapy. However, the Belgian technology assessment (Neyt 2011) analyzed and noted – as was discussed in considerable detail at the FDA panel meeting on July 20, 2011 – that one year mortality was in fact an absolute 12.7% higher for 90 patients who underwent TAVI compared to standard therapy in the PARTNER trial’s continued access study (a continuation of the PARTNER RCT). There are a number of reasons as to why this could have occurred, including the continued access patients being at higher overall risk of death due to extensive comorbidities, or that there is considerable variability in the original mortality estimates.
Of 179 patients assigned to TAVI in Cohort B, six did not receive a valve; several received more than one valve; and four underwent valve-in-valve procedures. Regarding these valve-in-valve cases, the FDA Executive Summary [page 29] cautioned that if TAVI “becomes commercially available, widespread use of the valve-in-valve technique might occur. While this only occurred four times in the Cohort B study, there have been many reports of valve-in-valve usage in Europe. Without any pre-clinical testing, and limited clinical data, the FDA is unable to draw conclusions regarding the short- and long-term safety of SAPIEN valve-in-valve implantation.” CMS notes that valve-in-valve has also been done in Europe. While the safety, effectiveness and durability of this procedure is unknown, it is currently occurring in clinical practice. Clearly, more evidence is needed while this practice continues to evolve.
Adverse events were not equal between the inoperable Cohort B groups. Strokes, vascular events, and major bleeding were higher in the TAVI group than the standard therapy group. Stroke occurred more frequently in the TAVI group than in the standard therapy group at both 30 days (6.7% versus 1.7%, p = 0.03) and at one year (10.6% versus 4.5%, p = 0.04). However, in Table 6 of the FDA Executive Summary (updated June 11, 2011), the number of strokes observed in the TAVI group and reported at the panel meeting were higher than those published by Leon, et al. (2010), i.e., 7.3% at 30 days and 11.2% at one year, pointing to the variability in current adverse event estimates.
Furthermore in Cohort B, in the ITT (intention-to-treat) population, the number of all neurological events (stroke and TIA) over the entire study period was more than three times higher in the TAVI group (N = 25, 14%) as compared to the control group (N = 8, 4.5%). A more detailed examination reveals that the eight neurological events observed in standard therapy controls over the entire study period included no TIAs. Rather, in control patients, there was one hemorrhagic stroke at eight months and seven ischemic/unclassified strokes: one after open AVR; four after BAV (five days, two weeks, two months, and six months); two in patients who only received medical management (one on the day of randomization, and another three days after randomization) (FDA Executive Summary).
The FDA executive summary also noted: “Only 14 control patients had optimal medical therapy without an interventional procedure throughout the trial. As mentioned above, two of these 14 patients had strokes within 3 days of randomization, but there were no further strokes. Fourteen additional patients had either open AVR or apico-aortic conduits. One of these 14 patients had a stroke on the day of surgery. There were no further strokes throughout the trial in the Control group. Therefore, the control group had minimal neurological events over 60 days after invasive procedures and there does not appear to be a continuing risk of neurological events. As a result, there is no evidence that the patients in this study were a high risk stroke population.”
Moreover, in Cohort B’s TAVI group, there were three TIAs (143 days in one patient; 386 and 831 days in a second patient), three intracranial hemorrhages (51, 136, and 151 days), three hemorrhagic strokes (two, 39, and 120 days), and 16 ischemic/unclassified strokes: one occurred after randomization and before device implantation; 10 of 16 were recognized within six days of implantation or attempted implantation; two of 16 occurred from 23-180 days (23 and 75 days); and three of 16 occurred late (361, 650, and 875 days). “This shows that 12/25 (48%) of the neurological events occurred > 30 days after the procedure – thus indicating a continued risk of neurological events with the device” (FDA Executive Summary 2011, page 23). The risk of neurologic events should be better defined for patient decision making.
TAVI was also associated with a higher incidence of major vascular complications at 30 days (16.2% versus 1.1%, p < 0.001), as compared with standard therapy; and in the summary prepared for the July 20, 2011 Circulatory System Devices Panel meeting, the FDA reported that half (55.9%) of the TAVI patients had serious adverse events relating to the access procedure. Based on their review of the Clinical Events Committee narratives, the FDA Executive Summary (page 24) noted that the most serious of these vascular complications included: aortic dissection (1 patient), iliac artery/distal aortic injury (17 patients), femoral artery injury (13 patients), pseudoaneurysm (2 patients), hematoma (6 patients) and unknown injury (2 patients). The TAVI patients do receive heparin during the procedure and are anticoagulated post-procedure, but it is not clear if bleeding events are attributable to this anticoagulation. More information on anticoagulation is needed for optimal patient management.
Several groups (Kahlert 2010, Ghanem 2010, Arnold 2010, Rodes-Cabau 2011, Astarci 2011) have also published series documenting the rate of clinically silent cerebral ischemia following TAVI. Kahlert and colleagues (2010), for instance, assessed peri-procedural apparent and silent cerebral ischemia by neurological testing and serial cerebral diffusion-weighted magnetic resonance imaging (DW-MRI) at baseline, 2.5 to 4.4 days after the procedure, and at three months after TAVI. Clinically silent new foci were detected in almost all patients (84%) undergoing TAVI in Kahlert’s series, which (while typically multiple) were not associated with apparent neurological events or measurable deterioration of neurocognitive function during three-month follow-up. The clinical significance of these findings is unclear, with further investigative work needing to be done to inform patient understanding of the procedure.
A paravalvular leak refers to aortic regurgitation occurring due to mismatching of the implanted prosthetic valve and native aortic annulus, such that during diastole, part of the forward blood flow into the ascending aorta flows back between the prosthesis and the annulus.
In Cohort B, moderate or severe paravalvular aortic regurgitation was present in 11.8% of TAVI patients at 30 days and in 10.5% of TAVI patients at one year. Additionally, when both central regurgitation and paravalvular leak were included, 15.6% of TAVI patients had moderate or severe aortic regurgitation at one year. The FDA Executive Summary (page 25) noted that this amount of aortic regurgitation was appreciable and did not decrease over time in the TAVI group, and that the degree of aortic regurgitation remains a concern which will need to be monitored in subsequent studies. Critically, the presence of moderate to severe aortic regurgitation has been recently shown to be an independent predictor of mortality (Moat 2011; Abdel-Wahab 2011).
Cohort A
While outcomes for Cohort A have been reported (Smith 2011), at the time of this national coverage analysis the data for this cohort remains under FDA review. Here also the primary endpoint is death from any cause at one year in the intention-to-treat population, and there was a nonsignificant difference that was within the trial’s non-inferiority margin (24.2% in the TAVR group versus 26.8% in the surgical AVR group) for PARTNER Cohort A. Notably, however, greater than 10% (N = 38) of the patients randomized to surgical replacement in Cohort A were not treated as assigned – mainly due to patient refusal or withdrawal, including 27/248 surgical replacement control patients in the transfemoral cohort and 11/103 surgical replacement control patients in the transapical cohort (N = 103). Only 4/244 (1.1%) patients in the transfemoral TAVR group were not treated as assigned by randomization. Recurrent hospitalization rates was a secondary endpoint in Cohort A, and at one year rehospitalization occurred in 58 patients (18.2%) in the TAVR group and in 45 (15.5%) in the surgical AVR group, a non-statistically significant difference. The functional outcomes of NYHA class and 6-minute walk showed a difference at 30 days, but at one year the earlier between-group differences were not evident. The one year QoL results for PARTNER’s Cohort A (while publicly presented) have yet to be published.
In Cohort A, adverse events were not equal between the TAVI group and the surgical AVR group. Strokes and vascular events were higher in the TAVI group than the surgical group, with major bleeding higher in the surgical group. Stroke was a pre-specified secondary end point, and all neurological events (comprising major stroke, minor stroke and TIA) occurred more frequently in the TAVR group than in the surgical AVR group, both at 30 days (5.5% versus 2.4%, p = 0.04) and at one year (8.3% versus 4.3%, p = 0.04). Major strokes (while not a pre-specified endpoint) likewise occurred more frequently in the TAVR group than in the surgical AVR group, both at 30 days (3.8% versus 2.1%, p = 0.20) and at one year (5.1% versus 2.4%, p = 0.07). Vascular complications and bleeding were additional pre-specified secondary safety endpoints. At 30 days, the TAVR group had a significantly higher rate of vascular complications than did the surgical AVR group (17.0% versus 3.8%, p<0.001); and the TAVR group had significantly higher rate of major vascular complications[1] than did the surgical group (11.0% versus 3.2%, p < 0.001).
Notably, in PARTNER Cohort A, moderate or severe paravalvular aortic regurgitation occurred more frequently in the TAVR group versus the surgical AVR group at both 30 days (12.2% versus 0.9%, p < 0.001) and at one year (6.8% versus 1.9%, p < 0.001). In Moat and colleagues’ (2011) all-inclusive TAVR registry series, follow-up ranged from 11-46 months; and mortality tracking was achieved in 100% of patients with survival status reported as of December 12, 2010. In data collected prospectively for Moat’s consecutive series of 870 high-risk patients undergoing 877 TAVR procedures (regardless of technology or access route) in all 25 centers undertaking TAVR in the United Kingdom, the presence of moderate to severe aortic regurgitation was an independent predictor of mortality. Likewise, in another recent large series evaluating post-procedural aortic regurgitation in 690 of 697 consecutive transcatheter aortic valve implantations, Abdel-Wahab (2011) found that significant (≥ 2/4) aortic regurgitation occurred in 119 patients (17.2%) and was a strong independent predictor of in-hospital death. More investigation is needed to understand paravalvular leaks and the effect on patient outcomes.
Finally, the spectrum of aortic stenosis is a continuum and some patients with severe symptomatic aortic stenosis are either too sick or have such severe comorbidities that, despite TAVI, these patients – the so-called “Cohort C” patients described in the FDA Executive Summary (page 29) – will not improve functionally or live longer following intervention:
“The FDA worked extensively with sponsor to define “inoperable” or “extreme high risk” for this randomized study of inoperable patients so as not to enroll less sick patients who could reasonably have open AVR. However, active consideration was not given to specifying patients who should not have transcatheter valve implantation due to extensive comorbidities. There were no specific inclusion/exclusion criteria in this study to eliminate patients too sick to benefit from isolated treatment of severe aortic stenosis.
Based on a review of the CEC narratives, it is clear that one needs also to consider when transcatheter valve implantation may not have a positive impact on a patient’s quality of life. In addition, SAPIEN implantation requires general anesthesia, 4+ hours of procedure time, radiographic contrast, invasive TEE, often an operative procedure for vascular access or closure, etc.; therefore, it is a highly invasive interventional cardiology procedure.”
Likewise of importance, more than death itself, many elderly patients fear loss of independence, becoming a burden to family, and/or nursing home admission. For such individuals considering surgical or transcatheter AVR, stroke may be worse than death.
The impact of comorbidities on potential benefit or harm from TAVR has not currently been established for patients with intra-cardiac mass, thrombus or vegetation; congenital unicuspid or bicuspid aortic valve; or contra-indication to anticoagulation medications. Also, the impact of those comorbidities may evolve over time with advances in clinical care. Accordingly, our final decision reflects flexibility to give physician researchers the ability to determine which patients will likely benefit, versus those who are too sick to benefit, from the procedure.
- Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for Medicare beneficiaries with severe symptomatic aortic stenosis who are not candidates for surgical aortic valve replacement? [PARTNER Cohort B]
For the highly selected patients in Cohort B, the evidence is not adequate to conclude that TAVR generally improves health outcomes for Medicare beneficiaries with severe symptomatic native aortic stenosis who were deemed not to be suitable candidates for surgical AVR. TAVR, however, may improve health outcomes in very highly selected, well-informed, inoperable patients when added safety and patient protections are in place in carefully monitored clinical studies performed by expert multi-disciplinary heart teams in facilities that furnish an appropriate environment, which can be available through CED under §1862(a)(1)(E) of the Act. We believe that the STS ACC TAVT Registry, as currently designed, is an appropriate platform for a carefully monitored clinical study for this purpose. We also believe that adherence to facility and practitioner criteria based on guidance developed jointly by AATS, ACCF, SCAI and STS and informed by public comment should be considered to satisfy the CMS requirements for facilities and practitioners.
-
Is the evidence adequate to conclude that transcatheter aortic valve replacement improves health outcomes for high surgical risk Medicare beneficiaries with severe symptomatic aortic stenosis who are candidates for surgical aortic valve replacement? [PARTNER Cohort A]
For the highly selected patients in Cohort A with symptomatic native aortic stenosis who were deemed candidates for surgical aortic valve replacement, TAVR provided no mortality benefit but significant risk of harms. TAVR, however, may yet be demonstrated to improve health outcomes in very highly selected, well-informed, operable patients seeking added safety and patient protection available in carefully monitored clinical studies performed by expert multi-disciplinary heart teams in facilities that furnish an appropriate environment under §1862(a)(1)(E) of the Act.
We believe that gaps in the current evidence base lead to uncertainty about the overall impact of TAVR on beneficiary outcomes when furnished outside of the setting of evidence development or clinical trial protocols. The following points describe some of our concerns.
- The STS risk score and EuroSCORE give operative risk information, but do not predict the important patient-centered outcome of quality of life improvement. For patient selection and informed consent, information about quality of life improvement as it applies to individual patient decision making should be available.
- Accurate risk prediction is important. There are no specific recommendations for defining inoperability so this depends on the judgment of the medical team. Assessment can vary and be dependent on surgeon and institutional experience. A clearer understanding of comorbid conditions that affect patient outcomes is crucial. Furthermore, the impact of “unmeasured covariates” that enter into “clinical judgment” is unknown and likely critical for patient outcomes (Sundt 2009). Better tools are needed to assist both physicians and patients in risk ascertainment.
- Mortality (all-cause) at 30 days was less in the randomized PARTNER trial data as compared to the SOURCE registry (consecutive patients in Europe after commercialization) (Thomas et al 2010). Therefore, it remains unclear if the randomized trial data that were generated under optimal procedural circumstances is generalizable to routine clinical practice.
- The clinical significance of clinically silent cerebral ischemia is unknown. An examination of both short and long term quality of life information is needed to inform patient understanding of this procedure.
- The assessment of treatment success should encompass the reasonably expected durability of the treatment and extend beyond the mere technical completion of the operative procedure.
- Mixed results in the evidence base to date may reflect differences that may be predictable. Utilizing the CED approach is important to ensure that future care is informed by lessons learned.
For example, in addition to critically evaluating each patient’s quality of life pre- and post-TAVR, future clinical research studies should most assuredly address:
- What are the determinants and impact of para-prosthetic leaks and paravalvular aortic regurgitation upon rates of death and stroke in TAVR patients as compared to non-TAVR controls?
- What is the echocardiographic, CT and/or MR assessment of transcatheter aortic valvular performance, deterioration and durability as compared to surgical AVR with a mechanical or bioprosthetic valve?
- Can a better frailty index be developed and validated to improve patient selection for TAVR?
- Based on pre- and post-procedure diffusion-weighted MR (DW-MRI), what is the influence of clinically silent strokes upon memory and neurocognition following TAVR as compared to non-TAVR controls?
- What represents optimal peri-procedural and post-procedural antiplatelet and anticoagulation therapy for TAVR?
Additionally, the learning curve with this complex technology appears substantial (Nuis 2011, Alli 2011). For instance, Gurvitch 2011 suggests that procedural experience is an independent predictor of 30-day mortality. In a recent study by Alli, they stated, “Our data show increasing proficiency with evidence of plateau after the first 30 cases. More studies are needed to confirm these findings.”(Alli 2011) In this cohort at the Mayo clinic, the 30-day mortality was 11%, clearly higher than the PARTNER results. The correlation of volume to mortality, and morbidity can be clarified with additional evidence.
The data used for the FDA PMA approval were generated under rigorous clinical trial conditions. To enhance the likelihood that beneficiaries experience similar improved health outcomes overall, operator and facility criteria are important and need definition. The formal request for this analysis broadly outlined desirable operator and facility criteria for performing TAVR. Our criteria, listed in section I of this decision memorandum, are informed by the information included in the formal request and subsequent information submitted to CMS by the requestors such as the Journal of the American College of Cardiology (JACC) entitled 2012 ACCF/AATS/SCAI/STS Expert Consensus Document on Transcatheter Aortic Valve Replacement (Holmes 2012) (http://content.onlinejacc.org/cgi/reprint/j.jacc.2012.01.001v1.pdf) as well as our own review of the evidence and public comment. As such, we are incorporating the operator and facility criteria that we believe are appropriate in section I of this decision.
In this rapidly evolving technology, the incidence of stroke and other adverse outcomes may decline with improvements in patient selection, device characteristics, and procedural practices. Monitoring these changes that ultimately lead to improvements in morbidity and mortality is critical.
Disparities in Transcatheter Aortic Valve Replacement
In the PARTNER Cohort B trial, 46% of study participants were men and 92% were Caucasian. Makary (2010) reported that frailty independently predicts postoperative complications, length of hospital stay, and discharge to a skilled or assisted-living facility in older surgical patients, and Zenilman (2011) noted that prevalence of frailty among those > 65 years old has been estimated at 7-16% and is more common among women and African Americans.
Summary
Upon review of the available evidence and public comments, we believe that the requestors’ arguments are generally persuasive and we believe that our decision to cover FDA indicated uses under CED (registry participation) is consistent with the formal request for coverage from the professional societies. The requestors presented reasonable and supportable arguments for restricting coverage for indicated uses to practitioners and facilities meeting specified criteria, the derivation of which is from a consensus among the professional societies, and for requiring ongoing data collection. We agree that robust and reasonable practitioner and facility criteria can be articulated with considerations of public comment and concerns for beneficiary access and that their presence will improve beneficiary outcomes. We believe that CMS criteria for data collection and analysis for FDA indications can be met through enrollment and participation in a national prospective TAVR registry. We also believe that this decision is consistent with the FDA requirement for continuing data collection and analysis.
We believe that there is promising but inadequate evidence to conclude at this time that TAVR generally improves health outcomes for Medicare beneficiaries with symptomatic aortic stenosis. We believe that the beneficiaries’ ability to attain improved health outcomes is maximized when TAVR is furnished in settings that reflect those in the pivotal PARTNER trial, by appropriately trained, experienced operators in the context of a multidisciplinary team in a setting that assures sufficient volume to maintain proficiency. We are also mindful of ongoing research and recognize that an alternative to open surgical aortic valve replacement may be clinically appropriate and preferable in selected patients when certain protections are in place to enhance the likelihood of benefit. We also believe that the additional data collected in the context of a clinical setting can further clarify the impact of TAVR on the health of this Medicare patient population. We believe that Medicare coverage under the Coverage with Study Participation CED paradigm balances these considerations in the interests of our beneficiaries.
It is not apparent to us at this time that the available evidence clearly distinguishes patients who will experience an improved outcome from those who will derive harm such as a stroke or death, especially beyond one year post TAVR. Given the availability of an effective treatment - open surgical valve replacement – we believe that additional evidence needs to be developed to better inform treatment decisions and for fully informed discussions of risk and benefit of TAVR in operable patients with symptomatic aortic stenosis. We believe that this evidence can be developed in the context of clinical studies that meet the criteria specified in section B of the decision.
There are inherent challenges in developing durable conclusions about an invasive technically complex surgical procedure when much of the non-trial experience has accrued in other countries. Though technical factors and underlying patient physiology would be expected to vary little among countries, the practice of medicine reasonably reflects cultural expectations and local incentives for the behaviors of patients and physicians that may not align with those factors in the United States.
The success of surgical procedures depends heavily on the skill and experience of the operator(s) and the supporting environment for the procedure itself and for postoperative care of the patient. We recognize, as the requestors have noted, that new technologies demonstrate a learning curve. This leads logically to caution about expecting that results achieved by selected experts working within the parameters of a formal clinical trial protocol will be seen when the technology is disseminated to less experienced operators in non-trial settings. Experience also tells us that, with disseminated use over time, adverse event signals may become stronger and more apparent than initial data have indicated. At the same time, we believe that reported health outcomes can improve over time as operators gain more training and experience and as the collective experience leads to improvements in the technology itself. Both of these are relevant to TAVR.
For patients with symptomatic native aortic stenosis in Smith and colleagues’ (2011) PARTNER Cohort A who were deemed to be high surgical risk for surgical AVR, transcatheter aortic valve replacement provided no mortality benefit but rather increased risk of harms, including both significantly increased rates of stroke at 30 days and one year, as well as significantly increased vascular complications at 30 days.
That is, while TAVR may at some time in the future reduce overall morbidity and mortality for a better defined subset of high surgical risk patients, such a result has not yet been conclusively demonstrated. Furthermore, adopting this potentially transformational technology for use in moderate or lower risk populations beyond the selected high surgical risk population studied in the PARTNER trial is not appropriate at the current time.
Furthermore, where frailty was assessed by quantifying ability of patients to perform activities of daily living, as well as by performing a hand grip and a walk test, frailty was more often present in the standard therapy controls than TAVI patients (28.0% versus 18%) in the PARTNER Cohort B. Such imbalance in both frailty as a baseline characteristic, as well as the overwhelming lack of racial diversity in the study participants who were enrolled, severely limits both the internal and external validity of the PARTNER Cohort B trial.
We have noted the absolute 12.7% increased mortality for TAVI as compared to standard therapy reported for the 90 randomized patients in PARTNER’s Continued Access study. Specifically, for patients with severe symptomatic native aortic stenosis in Leon and colleagues’ (2010) PARTNER Cohort B who were jointly deemed inoperable by a cardiologist and at least two cardiovascular surgeons and who then underwent transcatheter aortic valve implantation, the pivotal trial’s published mortality benefit of 20% – while promising for some patients who may not fear stroke more than death – is not generalizable beyond this very highly selected study population and may overestimate TAVI’s treatment effect – particularly when one considers the uneven distribution of baseline characteristics (especially atrial fibrillation, COPD and frailty) which were greater in the standard therapy controls. Moreover, in the PARTNER Cohort B, strokes were significantly more frequent in the TAVI group at both 30 days and at one year as compared with standard care; and TAVI was associated with significantly greater major vascular complications at 30 days as compared with standard therapy.
Critically, in addition to the notably high incidence of incompletely understood, clinically silent cerebral emboli detected by DW-MRI following TAVI, patients in Cohort B who underwent TAVI experienced two and a half times more strokes than those who did not receive an implanted valve; and nearly half (48%) of the neurological events (stroke and TIA) in Cohort B occurred more than 30 days after the procedure – which as noted by the FDA indicated a “continued risk of neurological events with the device.”
Overarching concerns for TAVI are therefore high incidence of paravalvular aortic regurgitation (rare in surgical AVR) and post-procedural strokes that are possibly related to embolic material from the device itself and/or the unopposed space (paravalvular leak) between the implanted valvular prosthesis and the native aortic annulus. Such serious sequelae would be magnified in younger patients and/or lower to moderate risk patient populations with fewer comorbidities and longer life expectancy.
Results of the PARTNER trial cannot be extrapolated beyond the very highly selected Cohort A and Cohort B study populations or beyond the expert multidisciplinary heart teams and specialized facilities utilized in the PARTNER trial. Therefore, we believe that Medicare beneficiaries are more likely to experience the best achievable outcomes when TAVR is furnished in a manner that replicates the safeguards contained in the PARTNER protocol. We recognize the distinction between protocol requirements that address the clinical delivery of the then-investigational item or service itself from those that address administrative aspects of clinical research. We believe this decision reflects an appropriately balanced consideration of the topic, mindful of beneficiary outcomes and clinical efficiency.
IX. Conclusion
We believe that TAVR may, upon the development of additional evidence, prove to represent a substantial benefit to Medicare beneficiaries with severe symptomatic aortic valve stenosis, especially those for whom open surgical aortic valve replacement would be contraindicated or high risk.
The Centers for Medicare & Medicaid Services (CMS) covers transcatheter aortic valve replacement (TAVR) under Coverage with Evidence Development (CED) with the following conditions:
TAVR is covered for the treatment of symptomatic aortic valve stenosis when furnished according to an FDA approved indication and when all of the following conditions are met.
-
The procedure is furnished with a complete aortic valve and implantation system that has received FDA premarket approval (PMA) for that system’s FDA approved indication.
- Two cardiac surgeons have independently examined the patient face-to-face and evaluated the patient’s suitability for open aortic valve replacement (AVR) surgery; and both surgeons have documented the rationale for their clinical judgment and the rationale is available to the heart team.
- The patient (preoperatively and postoperatively) is under the care of a heart team: a cohesive, multi-disciplinary, team of medical professionals. The heart team concept embodies collaboration and dedication across medical specialties to offer optimal patient-centered care.
TAVR must be furnished in a hospital with the appropriate infrastructure that includes but is not limited to:
- On-site heart valve surgery program,
- Cardiac catheterization lab or hybrid operating room/catheterization lab equipped with a fixed radiographic imaging system with flat-panel fluoroscopy, offering quality imaging,
- Non-invasive imaging such as echocardiography, vascular ultrasound, computed tomography (CT) and magnetic resonance (MR),
- Sufficient space, in a sterile environment, to accommodate necessary equipment for cases with and without complications,
- Post-procedure intensive care facility with personnel experienced in managing patients who have undergone open-heart valve procedures,
- Appropriate volume requirements per the applicable qualifications below.
There are two sets of qualifications; the first set outlined below is for hospital programs and heart teams without previous TAVR experience and the second set is for those with TAVR experience.
Qualifications to begin a TAVR program for hospitals without TAVR experience:
The hospital program must have the following:
- ≥ 50 total AVRs in the previous year prior to TAVR, including ≥ 10 high-risk patients, and;
- ≥ 2 physicians with cardiac surgery privileges, and;
- ≥ 1000 catheterizations per year, including ≥ 400 percutaneous coronary interventions (PCIs) per year.
Qualifications to begin a TAVR program for heart teams without TAVR experience:
The heart team must include:
- Cardiovascular surgeon with:
- ≥ 100 career AVRs including 10 high-risk patients; or
- ≥ 25 AVRs in one year; or
- ≥ 50 AVRs in 2 years; and which include at least 20 AVRs in the last year prior to TAVR initiation; and
- Interventional cardiologist with:
- Professional experience with 100 structural heart disease procedures lifetime; or;
- 30 left-sided structural procedures per year of which 60% should be balloon aortic valvuloplasty (BAV). Atrial septal defect and patent foramen ovale closure are not considered left-sided procedures; and
- Additional members of the heart team such as echocardiographers, imaging specialists, heart failure specialists, cardiac anesthesiologists, intensivists, nurses, and social workers; and
- Device-specific training as required by the manufacturer.
Qualifications for hospital programs with TAVR experience:
The hospital program must maintain the following:
- ≥ 20 AVRs per year or ≥ 40 AVRs every 2 years; and
- ≥ 2 physicians with cardiac surgery privileges; and
- ≥ 1000 catheterizations per year, including ≥ 400 percutaneous coronary interventions (PCIs) per year.
Qualifications for heart teams with TAVR experience:
The heart team must include:
- A cardiovascular surgeon and an interventional cardiologist whose combined experience maintains the following:
- ≥ 20 TAVR procedures in the prior year, or;
- ≥ 40 TAVR procedures in the prior 2 years; and
- Additional members of the heart team such as echocardiographers, imaging specialists, heart failure specialists, cardiac anesthesiologists, intensivists, nurses, and social workers.
- The heart team’s interventional cardiologist(s) and cardiac surgeon(s) must jointly participate in the intra-operative technical aspects of TAVR.
- The heart team and hospital are participating in a prospective, national, audited registry that: 1) consecutively enrolls TAVR patients; 2) accepts all manufactured devices; 3) follows the patient for at least one year; and 4) complies with relevant regulations relating to protecting human research subjects, including 45 CFR Part 46 and 21 CFR Parts 50 & 56. The following outcomes must be tracked by the registry; and the registry must be designed to permit identification and analysis of patient, practitioner and facility level variables that predict each of these outcomes:
- Stroke;
- All cause mortality;
- Transient Ischemic Attacks (TIAs);
- Major vascular events;
- Acute kidney injury;
- Repeat aortic valve procedures;
- Quality of Life (QoL).
The registry should collect all data necessary and have a written executable analysis plan in place to address the following questions (to appropriately address some questions, Medicare claims or other outside data may be necessary):
- When performed outside a controlled clinical study, how do outcomes and adverse events compare
to the pivotal clinical studies?
- How do outcomes and adverse events in subpopulations compare to patients in the pivotal
clinical studies?
- What is the long term ( ≥ 5 year) durability of the device?
- What are the long term ( ≥ 5 year) outcomes and adverse events?
- How do the demographics of registry patients compare to the pivotal studies?
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
TAVR is covered for uses that are not expressly listed as an FDA approved indication when performed within a clinical study that fulfills all of the following.
-
The heart team’s interventional cardiologist(s) and cardiac surgeon(s) must jointly participate in the intra-operative technical aspects of TAVR.
- As a fully-described, written part of its protocol, the clinical research study must critically evaluate not only each patient’s quality of life pre- and post-TAVR (minimum of 1 year), but must also address at least one of the following questions:
- What is the incidence of stroke?
- What is the rate of all cause mortality?
- What is the incidence of transient ischemic attacks (TIAs)?
- What is the incidence of major vascular events?
- What is the incidence of acute kidney injury?
- What is the incidence of repeat aortic valve procedures?
- The clinical study must adhere to the following standards of scientific integrity and relevance to the Medicare population:
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants’ health outcomes.
- The research study is well supported by available scientific and medical information or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it also must be in compliance with 21 CFR Parts 50 and 56. In particular, the informed consent includes a straightforward explanation of the reported increased risks of stroke and vascular complications that have been published for TAVR.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed as Medicare coverage requirements.
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject.
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria affect enrollment of these populations, and a plan for the retention and reporting of said populations on the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
- The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Re: TAVR CED
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
TAVR is not covered for patients in whom existing co-morbidities would preclude the expected benefit from correction of the aortic stenosis.
Appendix A: General Methodological Principles of Study Design
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of an illness or injury or to improve the functioning of a malformed body member. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to help ensure adequate numbers of patients are enrolled to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to which group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is the extent to which differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of which have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
- Randomized controlled trials
- Non-randomized controlled trials
- Prospective cohort studies
- Retrospective case control studies
- Cross-sectional studies
- Surveillance studies (e.g., using registries or surveys)
- Consecutive case series
- Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in which confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to which the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. We are interested in the results of changed patient management not just altered management. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. For most determinations, CMS evaluates whether reported benefits translate into improved health outcomes. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.
Appendix B: Tables
| Outcome |
30 Days [Cohort A] |
1 Year [Cohort A] |
| |
Transcatheter Replacement
(N=348) |
Surgical Replacement
(N=351) |
P Value |
Transcatheter Replacement
(N=348) |
Surgical Replacement
(N=351) |
P Value |
| Number of Patients (%) |
Number of Patients (%) |
| Death, from any cause |
12 (3.4) |
22 (6.5) |
0.07 |
84 (24.2) |
89 (26.8) |
0.44 |
| Death, from cardiac causes |
11 (3.2) |
10 (3.0) |
0.90 |
47 (14.3) |
40 (13.0) |
0.63 |
| Repeat hospitalization |
15 (4.4) |
12 (3.7) |
0.64 |
58 (18.2) |
45 (15.5) |
0.38 |
| Death or repeat hospitalization |
25 (7.2) |
33 (9.7) |
0.24 |
120 (34.6) |
119 (35.9) |
0.73 |
| Stroke or TIA |
19 (5.5) |
8 (2.4) |
0.04 |
27 (8.3) |
13 (4.3) |
0.04 |
| TIA |
3 (0.9) |
1 (0.3) |
0.33 |
7 (2.3) |
4 (1.5) |
0.47 |
| Stroke, minor |
3 (0.9) |
1 (0.3) |
0.34 |
3 (0.9) |
2 (0.7) |
0.84 |
| Stroke, major |
13 (3.8) |
7 (2.1) |
0.20 |
17 (5.1) |
8 (2.4) |
0.07 |
| Death, from any cause or major stroke |
24 (6.9) |
28 (8.2) |
0.52 |
92 (26.5) |
93 (28.0) |
0.68 |
| Myocardial infarction |
0 |
2 (0.6) |
0.16 |
1 (0.4) |
2 (0.6) |
0.69 |
| Vascular complication, any |
59 (17.0) |
13 (3.8) |
<0.001 |
62 (18.0) |
16 (4.8) |
<0.001 |
| Vascular complication, major |
38 (11.0) |
11 (3.2) |
<0.001 |
39 (11.3) |
12 (3.5) |
<0.001 |
| Acute kidney injury, Creatinine >3mg/dl (256 umol/liter) |
4 (1.2) |
4 (1.2) |
0.95 |
12 (3.9) |
8 (2.7) |
0.41 |
| Acute kidney injury, renal-replacement therapy |
10 (2.9) |
10 (3.0) |
0.95 |
18 (5.4) |
20 (6.5) |
0.56 |
| Major bleeding |
32 (9.3) |
67 (19.5) |
<0.001 |
49 (14.7) |
85 (25.7) |
<0.001
|
| Endocarditis |
0 |
1 (0.3) |
0.32 |
2 (0.6) |
3 (1.0) |
0.63 |
| New-onset atrial fibrillation † |
30 (8.6) |
56 (16.0) |
0.006 |
42 (12.1) |
60 (17.1) |
0.07 |
| New pacemaker |
13 (3.8) |
12 (3.6) |
0.89 |
19 (5.7) |
16 (5.0) |
0.68 |
* All percentages are Kaplan-Meier estimates at the specific time point and thus do not equal the number of patients divided by the total number in the study group.
† The presence of new-onset atrial fibrillation was determined in an electrocardiography core laboratory.
From Leon, et al. (NEJM October 21, 2010) Table 2 – Clinical Outcomes at 30 Days and 1 Year*
NA denotes not applicable, TAVI transcatheter aortic-valve implantation, and TIA transient ischemic attack.
| Outcome |
30 Days [Cohort B] |
1 Year [Cohort B] |
|---|
| |
TAVI
(N=179) |
Standard Therapy
(N=179) |
P Value† |
TAVI
(N=179) |
Standard Therapy
(N=179) |
P Value† |
| Number of Patients (%) |
Number of Patients (%) |
| Death, from any cause |
9 (5.0) |
5 (2.8) |
0.41 |
55 (30.7) |
89 (49.7) |
<0.001 |
| Death, from cardiovascular cause‡ |
8 (4.5) |
3 (1.7) |
0.22 |
35 (19.6) |
75 (41.9) |
<0.001 |
| Repeat hospitalization ƒ |
10 (5.6) |
18 (10.1) |
0.17 |
40 (22.3) |
79 (44.1) |
<0.001 |
| Death, from any cause or repeat hospitalization ƒ |
19 (10.6) |
22 (12.3) |
0.74 |
76 (42.5) |
126 (70.4) |
<0.001 |
| Stroke or TIA, all |
12 (6.7) |
3 (1.7) |
0.03 |
19 (10.6) |
8 (4.5) |
0.04 |
| TIA |
0 |
0 |
|
1 (0.6) |
0 |
1.00 |
| Stroke, minor |
3 (1.7) |
1 (0.6) |
0.62 |
4 (2.2) |
1 (0.6) |
0.37
|
| Stroke, major |
9 (5.0) |
2 (1.1) |
0.06 |
14 (7.8) |
7 (3.9) |
0.18 |
| Death, from any cause or major stroke |
15 (8.4) |
7 (3.9) |
0.12 |
59 (33.0) |
90 (50.3) |
0.001 |
| Myocardial infarction, all |
0 |
0 |
|
1 (0.6) |
1 (0.6) |
1.00 |
| Myocardial infarction, periprocedural |
0 |
0 |
|
0 |
0 |
|
| Vascular complications, all |
55 (30.7) |
9 (5.0) |
<0.001 |
58 (32.4) |
13 (7.3) |
<0.001 |
| Vascular complications, major |
29 (16.2) |
2 (1.1) |
<0.001 |
30 (16.8) |
4 (2.2) |
<0.001 |
| Acute kidney injury, Creatinine>3 ¶ |
0 |
1 (0.6) |
1.00 |
2 (1.1) |
5 (2.8) |
0.45 |
| Acute kidney injury, renal-replacement therapy║ |
2 (1.1) |
3 (1.7) |
1.00 |
3 (1.7) |
6 (3.4) |
0.50 |
| Major bleeding |
30 (16.8) |
7 (3.9) |
<0.001 |
40 (22.3) |
20 (11.2) |
0.007 |
| Balloon aortic valvuloplasty |
1 (0.6) ** |
2 (1.1) |
1.00 |
1 (0.6) |
66 (36.9) †† |
<0.001 |
| Repeat TAVI ‡‡ |
3 (1.7) |
NA |
|
3 (1.7) |
NA |
|
| Aortic-valve replacement |
0 |
3 (1.7) |
0.25 |
2 (1.1) ** |
17 (9.5) |
<0.001 |
| Endocarditis |
0 |
0 |
|
2 (1.1) |
1 (0.6) |
0.31 |
| New atrial fibrillation |
1 (0.6) |
2 (1.1) |
1.00 |
1 (0.6) |
3 (1.7) |
0.62 |
| New pacemaker |
6 (3.4) |
9 (5.0) |
0.60 |
8 (4.5) |
14 (7.8) |
0.27 |
†P values are for between-group comparisons of the frequency of the event at each time point.
‡ Deaths from unknown causes were assumed to be deaths from cardiovascular causes.
ƒ Repeat hospitalizations were included if they were due to aortic stenosis or complications of the valve procedure (e.g., TAVI).
¶ Patients who received renal-replacement therapy were not included
║Patients who received renal-replacement therapy after randomizations were included.
** One patient in the TAVI group did not receive TAVI (because of failed access) and subsequently underwent balloon aortic valvuloplasty, followed by aortic-valve replacement.
†† A total of 30 patients underwent a repeat balloon aortic valvuloplasty after the index balloon aortic valvuloplasty procedure that had been performed in the first 30 days after randomization, and 36 patients underwent a first balloon aortic valvuloplasty more than 30 days after randomization.
‡‡ Three patients underwent a repeat TAVI within 24 hours after the index TAVI procedure; four patients in the standard-therapy group who underwent TAVI at a nonparticipating site outside the United States are not
included here.
Appendix C
[1] 1) Any thoracic aortic dissection; 2) access site or access-related vascular injury leading to either death, need for significant blood transfusions (>3 units), unplanned percutaneous or surgical intervention, or irreversible end-organ damage; 3) distal embolization (non-cerebral) from a vascular source requiring surgery or resulting in amputation or irreversible end-organ damage: or 4) left ventricular perforation.


