To: Administrative File: CAG-00432R
From: Louis Jacques, MD
Director, Coverage and Analysis Group
Tamara Syrek Jensen, JD
Deputy Director, Coverage and Analysis Group
Jyme Schafer, MD, MS
Director, Division of Medical and Surgical Services
Kimberly Smith, MD, MS
Lead Medical Officer, Division of Medical and Surgical Services
Marie Casey, BSN, MPH
Lead Analyst, Division of Medical and Surgical Services
Subject: Proposed Coverage Decision Memorandum for Ventricular Assist Devices
Date: August 1, 2013
I. Proposed Decision
The Centers for Medicare & Medicaid Services (CMS) proposes the following changes to the current national coverage determination (NCD).
- Ventricular assist devices (VADs) for bridge to transplant (BTT)
- For the existing requirement that a patient is approved and listed as a candidate for heart transplant by a Medicare-approved heart transplant center, we clearly identify that the patient must be active on the waitlist maintained by the Organ Procurement and Transplantation Network.
- Remove the existing requirement that a “Medicare-approved heart transplant center should make every reasonable effort to transplant patients on such devices as soon as medically reasonable. Ideally, the Medicare-approved heart transplant centers should determine patient-specific timetables for transplantation, and should not maintain such patients on VADs if suitable hearts become available.”
- VADs for destination therapy (DT)
- The evidence is insufficient to support changes to our current patient selection criteria for coverage of a VAD as DT.
- The evidence is sufficient to conclude that VADs implanted in facilities that meet certain criteria improve health outcomes for Medicare beneficiaries. Facilities currently credentialed by the Joint Commission for placement of VADs as DT may continue as Medicare-approved facilities for a period of one year following the posting of the final decision memorandum. At the conclusion of this transition period, these facilities must be in compliance with the following criteria as determined by the credentialing organization. As of the effective date, new facilities, must meet the following criteria as a condition of coverage of this procedure as DT under section 1862(a)(1)(A):
- Beneficiaries receiving VADs for DT must be managed by an explicitly identified cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in shared decision making and to provide appropriate informed consent.
The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum, all of the following:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 VADs over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical and device-based management including VADs, and clinical competence in the management of patients before and after heart transplant.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
- Facilities must track patient outcomes including survival, adverse events (e.g. bleeding, infection, stroke, and device malfunction), functional status, and quality of life in a way that allows comparisons with other institutions and facilitates internal quality monitoring and improvement.
- Facilities must be credentialed by an organization approved by CMS.
- We propose to remove the separate requirement that hospitals have in place staff and procedures for appropriate informed consent as this requirement is encompassed in the above team definition.
- The evidence is sufficient to conclude that continuing required participation in the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) will not adequately address the outstanding evidentiary questions for VADs; therefore, INTERMACS participation is no longer required for VADs to be determined reasonable and necessary and CMS proposes to remove this requirement.
- We propose to allow organizations that have credentialing programs specific to VADs to apply to CMS to be designated as a credentialing organization for VAD facilities for DT. These programs must ensure that credentialed facilities meet the criteria outlined in the NCD and may include additional requirements necessary to operationalize these criteria or administer the credentialing program. Beyond this, any additional substantive credentialing standards not identified in the final NCD are not required for Medicare coverage.
The process for organizations to apply for CMS approval to be designated as a credentialing organization for VAD facilities for DT will be posted on our web site along with a list of approved credentialing organizations, approved standard versions, and credentialed facilities. http://www.cms.gov/Medicare/Medicare-General-Information/MedicareApprovedFacilitie/VAD-Destination-Therapy-Facilities.html#TopOfPage.
- We propose to add a statement that the NCD does not address coverage of VADs for right ventricular support, biventricular support, use in patients under the age of 18, or use in patients with complex congenital heart disease and that coverage for items and services under section 1862(a)(1)(A) in these situations will be made by local Medicare Administrative Contractors (MACs) within their respective jurisdictions.
CMS is seeking comments on our proposed decision. We will respond to public comments in a final decision memorandum, as required by §1862(l)(3) of the Social Security Act (the Act).
In addition to the proposed changes above, CMS is renumbering its VAD-related policies into a sub-section of section 20.9 (Artificial Hearts and Related Devices) of the NCD Manual. The sub-section (20.9.1) will be titled Ventricular Assist Devices. This is an administrative change only to make it easier for the public to read and understand the VAD policies. Section 20.9.1 will include the existing coverage of VADs for postcardiotomy, BTT, and DT.
The changes to the manual are reflected in Appendix C.
II. Background
The following acronyms are used throughout this document. For the readers convenience they are listed here in alphabetical order.
6-MWD - six-minute walk distance
6-MWT - six-minute walk test
ACC - American College of Cardiology
ACCF - American College of Cardiology Foundation
ACE - Angiotensin-converting enzyme inhibitor
ACGME - Accreditation Council for Graduate Medical Education
ACP - American College of Physicians
AHA - American Heart Association
ARB - angiotensin receptor blocker
BiVAD - biventricular assist device
BMI - body mass index
BSA - body surface area
BTC - bridge to candidacy
BTT - bridge to transplant
CAP - continued access protocol
CDC - Centers for Disease Control and Prevention
CMS - Centers for Medicare & Medicaid Services
CRT - Cardiac resynchronization therapy
DNV - Det Norske Veritas Healthcare Inc.
DT - destination therapy
EQ-5D - EuroQuol-5D
EQ-5D VAS - EuroQol-5D Visual Analog Scale
FDA - Food and Drug Administration
HFrEF - heart failure with reduced ejection fraction
HFSA - Heart Failure Society of America
HM II - HeartMate II Left Ventricular Assist System
HM VE - HeartMate Vented Electric Left Ventricular Assist System
HM XVE - HeartMate XVE
HRQOL - health-related quality of life
HRSA - Health Resources and Services Administration
HW VAS - HeartWare Ventricular Assist System
IABP - intraaortic balloon pump
ICD - implantable cardioverter defibrillator
INTERMACS - Interagency Registry for Mechanically Assisted Circulatory Support
ISHLT - International Society for Heart and Lung Transplantation
LVAD - left ventricular assist device
LVEF - left ventricular ejection fraction
KCCQ - Kansas City Cardiomyopathy Questionnaire
KCCQ CSS - Kansas City Cardiomyopathy Questionnaire Clinical Summary Score
KCCQ OSS - Kansas City Cardiomyopathy Questionnaire Overall Summary Score
MAC - Medicare Administrative Contractor
MCS - mechanical circulatory support
MCSD - mechanical circulatory support device
MEDCAC - Medicare Evidence Development and Coverage Advisory Committee
METS - metabolic equivalent task score
MLHFQ - Minnesota Living with Heart Failure Questionnaire
NCA - National Coverage Analysis
NCD - National Coverage Determination
NHLBI - National Heart, Lung and Blood Institute
NIH - National Institutes of Health
NYHA - New York Heart Association
OPTN - Organ Procurement and Transplantation Network
PMA - premarket approval
PROs - patient-reported outcomes
QOL - quality of life
REMATCH - Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure
UNOS - United Network for Organ Sharing
VAD - ventricular assist device
The scope of this national coverage analysis (NCA) includes a review of the evidence for patient selection and facility criteria for the use of durable VADs for end-stage congestive heart failure. Specifically, we review the evidence for whether it supports changes to our current criteria.
Heart failure is a condition in which the heart cannot pump blood adequately to meet the body's needs at rest or with exertion. Around 5.7 million people in the United States have heart failure with a higher prevalence at older ages. In the Framingham Heart Study, the incidence of heart failure doubled for each additional ten years of age (Lloyd-Jones, et al., 2002). In addition to age, which is an independent risk factor for heart failure, older adults often have additional risk factors such as high blood pressure, diabetes mellitus, coronary heart disease, tobacco use, and overweight/obesity and may have been exposed to these risk factors for many years.
When the heart fails to adequately pump blood, patients retain excess fluid and tissues do not get enough oxygen. This results in symptoms such as shortness of breath, swelling of the legs, and fatigue and causes substantial morbidity and, in the most serious circumstances, mortality. Heart failure leads to over one million hospitalizations each year and 20% of hospitalizations in persons over the age of 65 (Go, et al., 2013) (Jessup & Brozena, 2003). It causes over 55,000 deaths and contributes to at least 275,000 deaths annually in the United States (CDC, 2012) (Go, et al., 2013).
While there are objective measures of the severity of heart failure such as ejection fraction and cardiopulmonary exercise testing, care is most often driven by symptom-based classifications including the New York Heart Association (NYHA) classification, INTERMACS profiles, and the American Heart Association and American College of Cardiology (AHA/ACC) Stages of Heart Failure.
The NYHA classification is a subjective measure of the severity of heart failure symptoms which some have criticized as being unresponsive to change, having a high degree of interobserver variability, and providing the perspective of the doctor rather than the patient (Green, et al., 2000) (Miller & Guglin, 2013). The four NYHA classes include:
- Class I: Patients with cardiac disease but without resulting limitation of physical activity. Ordinary physical activity does not cause undue fatigue, palpitation, dyspnea or anginal pain.
- Class II: Patients with cardiac disease resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity results in fatigue, palpitation, dyspnea or anginal pain.
- Class III: Patients with cardiac disease resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes fatigue, palpitation, dyspnea or anginal pain.
- Class IV: Patients with cardiac disease resulting in inability to carry on any physical activity without discomfort. Symptoms of heart failure or the anginal syndrome may be present even at rest. If any physical activity is undertaken, discomfort increases.
INTERMACS profiles were recently developed to further classify patients with advanced NYHA class III and class IV heart failure into one of seven profiles (Stevenson, et al., 2009):
- Profile 1 - Critical cardiogenic shock: Patient with life-threatening hypotension despite rapidly escalating inotropic support, critical organ hypoperfusion, often confirmed by worsening acidosis and/or lactate levels.
- Profile 2 - Progressive decline: Patient with declining function despite intravenous inotropic support, may manifest with worsening renal function, nutritional depletion, or inability to restore volume balance. Also describes declining status in patients unable to tolerate inotropic therapy.
- Profile 3 - Stable but inotrope dependent: Patient with stable blood pressure, organ function, nutrition, and symptoms on continuous intravenous inotropic support (or a temporary circulatory support device or both), but demonstrating repeated failure to wean from support due to recurrent symptomatic hypotension or renal dysfunction.
- Profile 4 - Resting symptoms: Patient who can be stabilized close to normal volume status but experiences daily symptoms of congestion at rest or during activities of daily living. Doses of diuretics generally fluctuate at very high levels.
- Profile 5 - Exertion intolerant: Patient who is comfortable at rest and with activities of daily living but is unable to engage in any other activity, living predominantly within the house.
- Profile 6 - Exertion limited: Patient without evidence of fluid overload who is comfortable at rest, and with activities of daily living and minor activities outside the home but fatigues after the first few minutes of any meaningful activity.
- Profile 7 - Advanced NYHA class III: Patient who is clinically stable with a reasonable level of comfortable activity, usually able to walk more than a block. Has a history of previous decompensation but any decompensation requiring intravenous diuretics or hospitalization within the previous month should make this person a patient profile 6 or lower.
The AHA/ACC Stages emphasize the development and progression of heart failure ranging from Stage A (risk factors but no current cardiac abnormality) to Stage D (refractory heart failure, may be eligible for advanced treatments such as continuous inotropes, heart transplant, ventricular assist device placement (VAD), or end-of-life care) (Hunt, 2009).
Therapeutic interventions include modification of diet and lifestyle (such as restricting dietary sodium intake or increasing exercise) and medications including diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), aldosterone antagonists, beta-blockers, or digoxin. In the most severe cases, intravenous inotropic medications can be used. Inotropic medications do not reverse heart failure but may improve symptoms by making the heart beat stronger or reducing strain on the heart by reducing blood pressure. Device therapies include implantable cardioverter defibrillators (ICD) for patients at risk for sudden cardiac death; pacemakers and cardiac resynchronization devices for patients with abnormalities in the heart's electrical conduction system; and VADs which are the subject of this analysis.
Heart failure can be a progressive disease with increasing symptoms over time despite optimal medical management, though the time course is difficult to predict. Eventually, the heart fails completely and can no longer pump enough blood to sustain life. At this end-stage, eligible patients can undergo heart transplant; however, only around 2,000 heart transplants are performed annually in the United States (HRSA, 2012). In addition, older patients are often not eligible for heart transplant due to comorbid conditions which greatly the increase risk of poor outcomes.
A ventricular assist device (VAD), also referred to as a mechanical circulatory support (MCS) device, is a mechanical pump that can assist a damaged or weakened heart in pumping blood. It does not replace the heart like a heart transplant but instead is surgically connected to the failing right or left ventricle of the native heart and the aorta. If both ventricles are failing, sometimes two devices are implanted for biventricular support. The mechanical pump is outside the body for temporary devices used in the hospital. The pump is implanted in the abdomen or chest for devices which allow patient mobility and hospital discharge, known as durable devices. All devices require a driveline that goes from the pump to an external power source and control unit. Initial pumps were pulsatile, mimicking the pulsations of the native heart, but clinical use was limited by issues such as large pump size, high rates of adverse events, and poor device durability. Newer continuous-flow pumps have almost entirely replaced pulsatile pumps for longer-term use.
Patients who may be candidates for VAD implant undergo extensive clinical testing to ensure an adequate severity of heart failure but acceptable severity of comorbidities. This evaluation attempts to balance the benefits that might be achieved by VAD implant with the significant risks of the surgery and prolonged device support. Initially, VADs were used in the hospital as short-term support for patients with acute heart failure caused by temporary conditions such as infection or open heart surgery. With the development of smaller implantable pumps, patients could be ambulatory, discharged from the hospital, and supported on device for longer periods of time. These durable VADs were first introduced in patients on the heart transplant waitlist as a "bridge to transplant (BTT)" since the duration of support was intended to be finite. With heart transplants in limited supply and additional clinical experience gained, devices were subsequently implanted as "destination therapy (DT)" in patients ineligible for heart transplant who required permanent support. Patients who are neither BTT nor DT are referred to as bridge to decision or bridge to candidacy (BTC).
The use of VADs as a therapeutic tool has continued to evolve since our last national coverage analyses for BTT and DT. Based on this and ten years of mandatory registry participation, CMS has decided to review our current BTT and DT policies. This national coverage analysis reviews the available evidence for patient selection and facility criteria for the use of durable VADs for end-stage congestive heart failure.
III. History of Medicare Coverage
Bridge to Transplant
In 1996, CMS began covering VADs implanted as BTT at Medicare-approved heart transplant centers. In 2001, CMS covered implantation at sites other than Medicare-approved heart transplant centers.
Currently, devices are only covered if they have received approval from the FDA for BTT, are used according to the FDA-approved labeling instructions, and all of the following criteria are fulfilled:
- The patient is approved and listed as a candidate for heart transplant by a Medicare-approved heart transplant center; and
- The implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved heart transplant center under which the patient is listed prior to implantation of the VAD.
- The Medicare-approved heart transplant center should make every reasonable effort to transplant patients on such devices as soon as medically reasonable. Ideally, the Medicare-approved heart transplant centers should determine patient-specific timetables for transplant, and should not maintain such patients on VADs if suitable hearts become available.
Destination Therapy
In 2003, CMS began covering VADs implanted as DT at Medicare-approved heart transplant centers meeting specific facility criteria including participation in a national, audited registry. We believed these criteria were necessary due to the technical nature of the procedure, the high-risk patient population, and the need to ensure reasonable dissemination of new technology. In 2007, CMS allowed implantation at sites other than Medicare-approved heart transplant centers, named INTERMACS as the required registry, and required facilities to be credentialed by the Joint Commission based on standards dated February 2007. INTERMACS is a North American registry of VAD recipients "established as a joint effort of the National Heart, Lung and Blood Institute (NHLBI), the Centers for Medicare and Medicaid Services (CMS), the Food and Drug Administration (FDA), clinicians, scientists and industry representatives" (http://www.uab.edu/intermacs/).
Currently, devices are only covered if they have received approval from the FDA for DT, are used according to the FDA-approved labeling instructions, and are implanted at a facility meeting the following criteria:
- Facilities must have at least one member of the VAD team with experience implanting at least 10 VADs (as bridge-to-transplant or destination therapy) or artificial hearts over the course of the previous 36 months;
- Facilities must be a member of the INTERMACS Registry; and,
- All facilities must meet the above facility criteria and be credentialed by the Joint Commission under the Disease Specific Certification Program for Ventricular Assist Devices (standards dated February 2007).
- Facilities also must have in place staff and procedures that ensure that prospective VAD recipients receive all information necessary to assist them in giving appropriate informed consent for the procedure so that they and their families are fully aware of the aftercare requirements and potential limitations, as well as benefits, following VAD implantation.
In 2003, CMS required patients implanted as DT to meet specific criteria which were developed based on the study that led to FDA approval of a pulsatile-flow device for DT (the pivotal study) (Rose, et al., 2001). In 2010, CMS modified these criteria after review of the pivotal study that led to FDA approval of a continuous-flow device for DT (Slaughter, et al., 2009).
Currently devices are only covered as DT for patients with chronic end-stage heart failure (New York Heart Association Class IV end-stage left ventricular failure) who are not candidates for heart transplant, and meet all of the following conditions:
- Have failed to respond to optimal medical management (including beta-blockers and ACE inhibitors if tolerated) for at least 45 of the last 60 days, or have been balloon pump-dependent for 7 days, or IV inotrope-dependent for 14 days; and
- Have a left ventricular ejection fraction (LVEF) < 25%; and
- Have demonstrated functional limitation with a peak oxygen consumption of < 14 ml/kg/min unless balloon pump- or inotrope-dependent or physically unable to perform the test.
A. Current Reconsideration
On February 7, 2013, CMS accepted a formal request from Det Norske Veritas Healthcare Inc. (DNV) to reconsider Section 20.9 of the NCD Manual. Specifically, DNV requests that the facility criteria be amended to include the DNV Mechanical Circulatory Support Certification Program as an acceptable credential for facilities implanting devices as DT. The request is available at http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/id268.pdf. CMS has also included a review of the existing evidence for patient selection and facility criteria for the use of durable VADs for end-stage congestive heart failure.
B. Benefit Category
For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act (the Act). VADs may fall within the Inpatient Hospital Services benefit category (section 1861(b) (2) of the Social Security Act (the Act)), which describes supplies, appliances, and equipment furnished by the hospital, for use in the hospital, for the care and treatment of inpatients. After a VAD has been surgically implanted into the patient and when the patient is not a hospital patient, the replacement of an external part or parts may be covered under Medicare Part B within the Prosthetic Device benefit category (section 1861(s)(8) of the Act).
This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
IV. Timeline of Recent Activities
| Date |
Action |
| February 7, 2013 |
CMS initiates this national coverage analysis. |
| March 9, 2013 |
Initial 30-day public comment period closes. |
V. FDA Status
The FDA is aware of several durable VADs which are currently FDA-approved and commercially marketed for use as BTT and/or DT for single-ventricle support in adults. The same VAD may also be FDA-approved for additional Indications for Use which will not be discussed here.
The FDA approved the pulsatile HeartMate Vented Electric Left Ventricular Assist System (HM VE) for BTT in 1994 and the modified HM XVE for DT in 2003 (Premarket Approval (PMA) P920014). This device has since been replaced by the continuous-flow HeartMate II Left Ventricular Assist System (HM II) which received FDA approval for BTT in 2008 and DT in 2010 (PMA P060040; http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P060040).
The HM II Summary of Safety and Effectiveness Indications for Use states:
"The HeartMate II LVAS is intended for use as a bridge to transplantation in cardiac transplant candidates at risk of imminent death from non-reversible left ventricular failure. The HeartMate II LVAS is also indicated for use in patients with New York Heart Association (NYHA) Class IIIB or IV end-stage left ventricular failure who have received optimal medical therapy for at least 45 of the last 60 days, and who are not candidates for cardiac transplantation."
The FDA approved the continuous-flow HeartWare Ventricular Assist System (HW VAS) for BTT in 2012 (PMA P100047; http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cftopic/pma/pma.cfm?num=P100047).
The HW VAS Summary of Safety and Effectiveness Indications for Use states:
"The HeartWare Ventricular Assist System (HeartWare VAS) is indicated for use as a bridge to cardiac transplantation in patients who are at risk of death from refractory end stage left ventricular heart failure."
VI. General Methodological Principles
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public commenters sometimes cite the published clinical evidence and provide CMS with useful information. Public comments that provide information based on unpublished evidence, such as the results of individual practitioners or patients, are less rigorous and, therefore, less useful for making a coverage determination. CMS uses the initial comment period to inform its proposed decision. CMS responds in detail to the public comments that were received in response to the proposed decision when it issues the final decision memorandum.
VII. Evidence
A. Introduction
This examination of evidence focuses upon whether new clinical evidence from peer-reviewed published literature or INTERMACS data analyses supports a change to our current NCD's patient selection criteria or facility criteria. Key health outcomes of interest to CMS are mortality; morbidity including adverse events such as bleeding, infection, stroke, or device malfunction; and patient-centered measures of physical function and quality of life (QOL).
Mortality in VAD recipients is generally reported using the Kaplan-Meier method. With this statistical technique, patients are censored at the time of VAD removal for heart transplant or recovery of native heart function and, therefore, their outcomes following device removal are not incorporated into subsequent data points.
Measures of patient function in heart failure include the NYHA classification, INTERMACS profiles, and the AHA/ACC Stages of Heart Failure which were discussed in section II. Additional measures include the six-minute walk test (6-MWT) and the metabolic equivalent task score (METS). The 6-MWT measures the distance a patient walks in six minutes (the six-minute walk distance; 6-MWD) (American Thoracic Society, 2002). While the testing is standardized, results are effort dependent. METS are a subjective rating of a patient's highest self-reported activity level during the reporting period ranging from 1 (bedridden, unable to care for self or participate in any physical activity) to 6 (dancing, climbing stairs, heavy shoveling).
Several instruments are available to assess QOL (Grady, et al., 2012) (Dunderdale, et al., 2005). Generic instruments such as the SF-36 or EuroQol-5D (EQ-5D) provide a broad overview and allow comparisons with healthy populations or those with other chronic conditions. Heart failure-specific instruments such as the Kansas City Cardiomyopathy Questionnaire (KCCQ) and Minnesota Living with Heart Failure Questionnaire (MLHFQ) are more relevant to the specific disease and may be more responsive to change but do not allow comparisons with other groups. No instruments have been designed or validated specifically for VAD recipients; however, the EQ-5D, KCCQ, and MLHFQ are most commonly reported. The EQ-5D scores patient-reported health-related quality of life (HRQOL) across different dimensions (mobility, self-care, usual activities, pain/discomfort, anxiety/depression). A higher value on a scale of 0 to 100 represents better quality of life. The KCCQ quantifies physical limitations, symptoms, self-efficacy, social limitations, and quality of life. The KCCQ Overall summary score (OSS) combines scores from all domains while the clinical summary score (CSS) combines the physical function and symptom scores. For both, a higher value on a range of 0 to 100 represents a better quality of life. The MLHFQ quantifies the impact of heart failure and its treatment on physical and emotional domains. A lower value on range of 0 to 105 represents a better quality of life.
Well-designed, double-blind randomized controlled trials provide the highest quality of evidence regarding patient outcomes as subjects are allocated to comparison groups in an unbiased way and outcomes are assessed without knowledge of the treatment. Our search did not identify any randomized trials reported since the last analyses for BTT and DT; therefore, we expanded our search to include study types lower in the evidence hierarchy such as non-randomized trials or observational cohorts.
B. Discussion of Evidence
1. Questions:
The development of an assessment in support of Medicare coverage decisions is based on the same general question for almost all national coverage analyses (NCAs): "Is the evidence sufficient to conclude that the application of the item or service under study will improve health outcomes for Medicare patients?" For this NCD, the specific questions of interest are:
1. Is the evidence adequate to conclude that maintaining the current patient selection criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
2. Is the evidence adequate to conclude that maintaining the current facility criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
2. External Technology Assessment (TA)
CMS did not commission an external TA for this NCA; however, we identified one external TA published since the last NCA.
Rector TS, Taylor BC, Greer N, Rutks I, and Wilt TJ. Use of Left Ventricular Assist Devices as Destination Therapy in End-Stage Congestive Heart Failure: A Systematic Review. VA-ESP Project #09-009; May 2012. (Rector, et al., 2012)
The Department of Veteran's Affairs Health Services Research & Development Service Evidence-based Synthesis Program performed a systematic review on the use of VADs as DT in end-stage congestive heart failure. They reviewed articles published through October 2011 addressing three key questions, one of which is relevant to the scope of this NCA.
"Key Question #2. What patient or site characteristics have been associated with patient benefits or harms when the FDA-approved, current generation LVAD is used as destination therapy?
Conclusion: The available evidence is insufficient to refine patient or site selection criteria for use of the HeartMate II as destination therapy.
A few studies have identified risk factors for mortality and complications and developed or applied mortality prediction models to this particular patient population. Further studies are needed to validate use of different criteria to improve patient outcomes. An ongoing clinical trial is selecting less severely ill patients and may expand the criteria for use of a newer generation continuous flow device (HeartWare) as destination therapy. In the meantime, the approved FDA indication and CMS criteria for coverage are available to guide patient selection."
The authors noted that "Patients who die in the hospital soon after implantation of a ventricular assist device do not benefit. A validated prediction model for early/postoperative mortality could be applied to avoid high risk and costly attempts to use ventricular assist devices as destination therapy. Ideally clinical trials would be done to show that use of an outcome prediction model improves patient outcomes. This review did not find any established or proposed threshold for predicted risk of post-operative mortality that would preclude use of destination therapy or generally be acceptable to patients and health care providers."
3. Internal Technology Assessment
CMS examined the evidence regarding the impact of VADs on mortality, morbidity, QOL, and functional status in patients with end-stage congestive heart failure. Specifically, we assessed whether the evidence is adequate to support changes to our current patient selection or facility criteria. We included studies with publication dates between August 1, 2007 and March 28, 2013 to include the first pivotal trial for FDA approval of a continuous-flow device and later studies. For DT, we included studies with publication dates between January 2010 and March 28, 2013 to only include studies published since our last NCA.
We searched the PubMed database using the terms ventricular assist device, mechanical circulatory support device, or INTERMACS. We limited our search to English language publications in humans over the age of 18. We reviewed the titles and abstracts of peer-reviewed publications and all potentially relevant articles were reviewed in full. We identified additional references from the bibliographies of key articles.
We excluded studies of devices not currently approved by the FDA, devices that are no longer marketed, pre-pivotal studies of approved devices, studies of fewer than 50 VAD recipients, retrospective single-center case series, studies reporting only intermediate or surrogate outcomes, studies only reporting outcomes following heart transplant, or studies of cost or cost effectiveness. We also excluded studies focused only on pulsatile, temporary (non-durable), percutaneous, right-sided, biventricular, or partial-support devices and studies of artificial hearts.
We identified three trials of the use of continuous-flow VADs meeting the above criteria: the pivotal HM II BTT trial (Miller, et al., 2007) (Pagani, et al., 2009) (Bogaev, et al., 2011), follow-up analyses of the pivotal HM II DT trial (initial trial reviewed previously in CAG-00119R2) (Park, et al., 2012), and the pivotal HW VAS BTT trial (Aaronson, et al., 2012). We identified two studies reporting additional analyses of data from these trials and their associated continued access protocols (CAP) (Adamson, et al., 2011) (Cowger, et al., 2013).
Additionally, we identified several published analyses of INTERMACS registry data: an FDA-required post-approval study of the HM II as BTT (Starling, et al., 2011), a retrospective analysis of HM II BTT implants (John, et al., 2011), a comparison of outcomes in men and women (Hsich, et al., 2012), and a comparison of device durability (Holman, et al., 2013). We also identified published (Kirklin, et al., 2012) (Kirklin, et al., 2013) and unpublished (INTERMACS Q4, 2012) (INTERMACS CMS Q4, 2012) (INTERMACS CMS Report, 2013) reports from the INTERMACS investigators.
We identified one study of a program intervention (Pamboukian, et al., 2011). Lastly, we identified one systematic review of the literature on patient-reported outcomes for VADs (Brouwers, et al., 2011).
Question #1: Is the evidence adequate to conclude that maintaining the current patient selection criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
We note that the studies identified to inform this first question are limited to pivotal trials which led to FDA device approvals, follow-up analyses of pivotal trial data, and INTERMACS analyses. Some of the INTERMACS analyses are not in the peer-reviewed published literature. We have reported a focused review of the population studied (e.g. inclusion/exclusion criteria) and their outcomes, specifically mortality, morbidity (adverse events), functional status, and QOL.
Miller LW, Pagani FD, Russell SD, John R, Boyle AJ, Aaronson KD, Conte JV, Naka Y, Mancini D, Delgado RM, MacGillivray TE, Farrar DJ, Frazier OH; Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med. 2007 Aug 30; 357(9): 885-96. PMID: 17761592. (Miller, et al., 2007)
Pagani FD, Miller LW, Russell SD, Aaronson KD, John R, Boyle AJ, Conte JV, Bogaev RC, MacGillivray TE, Naka Y, Mancini D, Massey HT, Chen L, Klodell CT, Aranda JM, Moazami N, Ewald GA, Farrar DJ, Frazier OH. Extended mechanical circulatory support with a continuous-flow rotary left ventricular assist device. J Am Coll Cardiol. 2009 Jul 21;54(4):312-21. PMID: 19608028. (Pagani, et al., 2009)
Miller et al. and Pagani et al. reported the results of a pivotal, manufacturer-sponsored, single-arm, prospective study and CAP which assessed outcomes for the HM II in a BTT population. Miller et al. reported results from 133 patients enrolled in the initial study. Pagani et al. reported follow-up outcomes for these patients combined with an additional 148 patients enrolled through a CAP.
Patients had NYHA class IV heart failure and were listed for heart transplant with United Network for Organ Sharing (UNOS) status 1A or 1B. Patients were excluded for severe renal, pulmonary, or hepatic dysfunction; active uncontrolled infection; a mechanical aortic valve; aortic insufficiency; an aortic aneurysm; the presence of other mechanical circulatory support, except for an intraaortic balloon pump (IABP); and technical obstacles thought by the investigator to pose an increased surgical risk. Ninety percent of patients were receiving intravenous inotropes with the remaining 10% intolerant due to arrhythmias. Forty-five percent were on an IABP.
Of the 281 total patients, 222 (79%) had either undergone heart transplant (n = 157), device explant for cardiac recovery (n = 7), or remained alive with ongoing mechanical support (n = 58) at 18 months. Kaplan-Meier survival for patients continuing on device support was 82% at six months (number at risk = 133) and 72% at 18 months (number at risk = 58).
Adverse event rates were compared with previous studies of pulsatile devices and included bleeding requiring surgery (0.45 events/patient-year vs. 1.47), driveline infection (0.26 vs. 3.49), stroke (0.14 vs. 0.44), non-stroke neurological events (0.09 vs. 0.67), and right heart failure requiring a right-sided VAD (0.09 vs. 0.30) (Frazier, et al., 2001). The authors noted, "differences in rates of adverse events may have been influenced by differences in acuity of patient illness or improvements in patient management over time."
From the initial 133 patients, 82 remained alive and on device support at the three month assessment of functional status and QOL. All reported NYHA class IV symptoms prior to implant. By three months (number tested =78) this fell to 3% with 32% reporting class I symptoms, 51% class II, and 14% class III. The number of patients able to perform the 6-MWT increased from 25 to 56 at three months and the mean 6-MWD increased from 42 ± 97 meters to 292 ± 212 meters. The mean score on the MLHFQ fell from 73 ± 25 (n = 114) to 45 ± 25 (n = 77). The mean KCCQ OSS rose from 33 ± 19 (n = 113) to 57 ± 20 (n = 77). Data collection was reportedly incomplete "because of issues related to staff availability, scheduling, or oversight."
Pagani et al. concluded, "A continuous-flow rotary pump LVAD with axial design provides safe, reliable, and effective hemodynamic support in patients awaiting transplantation with improved quality of life and functional capacity. Furthermore, LVAD therapy with continuous-flow rotary pumps with extended support is associated with a very low rate of device malfunction or infection requiring device exchange. Continuous-flow rotary pumps provide a superior alternative to pumps with a pulsatile design in patients awaiting transplantation."
Bogaev RC, Pamboukian SV, Moore SA, Chen L, John R, Boyle AJ, Sundareswaran KS, Farrar DJ, Frazier OH; Comparison of outcomes in women versus men using a continuous-flow left ventricular assist device as a bridge to transplantation. J Heart Lung Transplant. 2011 May; 30(5): 515-22. PMID: 21257321. (Bogaev, et al., 2011)
Bogaev et al. reported on "a sex-based analysis of the combined 465 patients who have received the HeartMate II as a bridge to cardiac transplantation." Specifically, they compared outcomes of 104 women and 361 men implanted with the HM II device during the BTT trial and CAP who either completed study endpoints or reached 18-month follow-up after implant.
Kaplan-Meier survival and adverse event rates were similar, but hemorrhagic stroke occurred more frequently in women (0.10 events/patient-year vs. 0.04) and device-related infections occurred less frequently (0.23 vs. 0.44).
The authors concluded, "continuous-flow LV assistance as a bridge to transplantation is associated with similar survival rates in women and men. Differences observed in higher stroke rates and fewer infections among women require further study."
Aaronson KD, Slaughter MS, Miller LW, McGee EC, Cotts WG, Acker MA, Jessup ML, Gregoric ID, Loyalka P, Frazier OH, Jeevanandam V, Anderson AS, Kormos RL, Teuteberg JJ, Levy WC, Naftel DC, Bittman RM, Pagani FD, Hathaway DR, Boyce SW. "Use of an intrapericardial, continuous-flow, centrifugal pump in patients awaiting heart transplantation." Circulation. 2012 Jun 26; 125(25): 3191-200. PMID: 22619284. (Aaronson, et al., 2012)
Aaronson et al. reported the results of a pivotal, manufacturer-sponsored, non-randomized, prospective noninferiority trial comparing outcomes of 140 patients implanted with the HW VAS as BTT with outcomes of a contemporaneous INTERMACS registry control group (>95% HM II).
HW VAS patients were ≥ 18 years of age with a body surface area (BSA) of ≥ 1.2 m2, had NYHA class IV symptoms, and were listed for heart transplant with UNOS status 1A or 1B. Patients were excluded for ongoing mechanical circulatory support with the exception of an IABP, a history of heart transplant, prior valve replacement, cirrhosis, portal hypertension, pulmonary hypertension unresponsive to medical management, untreated aortic aneurysm, symptomatic cerebrovascular disease or >80% carotid stenosis, severe right ventricular failure, active uncontrolled infection, thrombocytopenia, coagulopathy, intolerance to anticoagulant or antiplatelet therapy, serum creatinine greater than three times upper limit of normal or requirement for dialysis, liver enzymes greater than three times upper limit of normal, or recent cardiothoracic surgery, acute myocardial infarction, ventilator support, or pulmonary embolus. 95% of patients had NYHA class IV symptoms, 82% were on intravenous inotropes, and 25% of were on an IABP.
The INTERMACS control group included 499 adult patients who received a primary left-sided device during the study period, were prospectively enrolled in the registry, had a BSA of ≥ 1.2 m2, and were listed for heart transplant. Patients were excluded for a creatinine > 5 mg/dl, dialysis, or ventilator support within 24 hours of implant.
Ninety and seven tenths percent of HW VAS and 90.1% of control patients reached the primary endpoint of the proportion of patients who, at 180 days, had undergone transplant, had cardiac recovery, or remained on mechanical support with the originally implanted device. One year Kaplan-Meier survival was 86% for the HW VAS (number at risk = 63) and 85% for the INTERMACS control (number at risk = 186).
Adverse event rates for the HW VAS were generally comparable to the published literature (INTERMACS control data were unavailable). The authors noted that any adverse event comparisons were "solely hypothesis generating unless confirmed in a randomized clinical trial."
QOL was measured with the KCCQ and EQ-5D VAS. Ninety-one percent of patients completed the KCCQ at baseline and 76% of patients alive on device support completed it at six months. Available data demonstrated improvement in the KCCQ OSS from 35 ± 19 to 67 ± 21 and in the KCCQ CSS from 44 ± 22 to 74 ± 21. Ninety-three percent of patients completed the EQ-5D VAS at baseline and 78% of patients alive on device support completed it at six months. Available data demonstrated improvement in the EQ-5D VAS from 40 ± 24 to 70 ± 20.
Functional capacity was measured using the NYHA classification and 6-MWT. Changes in NYHA class were not reported due to significant missing data. Ninety-four percent of patients had a 6-MWD recorded at baseline and 80% of patients alive on device support had one recorded at six months. Available data demonstrated an improvement in median 6-MWD from 0 to 274.2 meters.
The authors concluded, "a small, continuous-flow, centrifugal pump with a single magnetically and hydrodynamically levitated moving part, implanted directly in the left ventricle and positioned within the pericardial space, was associated with high rates of 180-day success and survival and a favorable adverse event profile when used as a bridge to transplantation. Perioperative mortality was 1%, and survival at 1 year was 86%. Quality-of-life and functional capacity improvements were much larger than those seen with any drug or device therapy for advanced heart failure and were similar to those obtained with cardiac transplantation."
Park SJ, Milano CA, Tatooles AJ, Rogers JG, Adamson RM, Steidley DE, Ewald GA, Sundareswaran KS, Farrar DJ, Slaughter MS. Outcomes in advanced heart failure patients with left ventricular assist devices for destination therapy. Circ Heart Fail. 2012 Mar 1; 5(2):241-8. PMID: 22282104. (Park, et al., 2012)
Park et al. reported extended follow-up for 133 patients who received the HM II device during the pivotal HM II DT trial ("early-trial") compared with 281 patients enrolled in the CAP ("mid-trial") who had reached two-year follow-up. The authors stated, "The goal of this report is to compare outcomes in patients enrolled later in the trial under continued access protocol with outcomes of the initial primary patient cohort. The main hypothesis is that patients implanted in the later part of the trial would have better clinical outcomes compared with those who were implanted earlier."
We previously reviewed the pivotal HM II DT trial (CAG-00119R2) (Slaughter, et al., 2009). In brief, patients were enrolled with advanced heart failure refractory to medical management who were ineligible for heart transplant. Patients had an ejection fraction of < 25%, a peak VO2 < 14 mL/kg per minute or < 50% of predicted, NYHA Class IIIB or IV symptoms for at least 45 of 60 days, or dependence on an IABP for seven days or inotropes for 14 days before enrollment. Exclusion criteria included severe renal impairment (serum creatinine > 3.5 mg/dl or dialysis), hepatic or pulmonary dysfunction, uncontrolled infection, history of stroke, mechanical aortic valve, irreparable aortic insufficiency, aortic aneurysm > 5.0 cm, or other mechanical circulatory support (except IABPs). For this follow-up analysis, 71% of early-trial and 63% of mid-trial patients had NYHA class IV symptoms, 77% and 78% were receiving intravenous inotropes, and 23 and 19% were on an IABP respectively.
Kaplan-Meier survival in the early-trial group was 68% at one year (number at risk = 82) and 58% at two years (number at risk = 62) and in the mid-trial group survival was 73% (number at risk = 187) and 63% respectively (number at risk = 146).
Adverse events rates were compared between the early and mid-trial periods including hemorrhagic strokes (0.07 events/patient-year vs. 0.03), bleeding requiring transfusions (1.66 vs. 1.13), device-related infection (0.47 vs. 0.27), and sepsis (0.38 vs. 0.27).
QOL was measured using the KCCQ and MLHFQ and functional status was measured using the NYHA classification and 6-MWT. KCCQ results were available for 86% of early-trial and 87% of mid-trial patients at baseline with diminishing percentages available at later time points and similar degrees of missing data for the MLHFQ, NYHA class, and 6-MWT. The authors stated, "Early and sustained improvements in quality of life were seen in both groups, and there was a trend toward patients having a better quality of life in the Mid Trial group compared with Early Trial... Significant improvements in functional status over time were observed in both the Early and Mid-Trial groups"
The authors concluded, "The benefit of DT therapy with the HM II is confirmed in subsequent trial patients. The survival rates in these patients are now 73% at 1 year and 63% at 2 years. These were substantial reductions in serious adverse events including hemorrhagic strokes (> 50% reduction), localized non-device-related infection (35% reduction), sepsis (30% reduction), device-related infections (> 40% reduction), bleeding requiring transfusion (> 30% reductions), and cardiac arrhythmias (> 30% reduction). There were also fewer deaths due to hemorrhagic strokes. These improvements highlight that both clinicians and patients are benefiting from the increasing clinical experience associated with the use of HM II for long-term treatment of advanced heart failure, which are directly getting translated to improving clinical outcomes."
Adamson RM, Stahovich M, Chillcott S, Baradarian S, Chammas J, Jaski B, Hoagland P, Dembitsky W. Clinical strategies and outcomes in advanced heart failure patients older than 70 years of age receiving the HeartMate II left ventricular assist device: a community hospital experience. J Am Coll Cardiol. 2011 Jun 21; 57(25): 2487-95. PMID: 21679851. (Adamson, et al., 2011)
Adamson et al. reported the outcomes of 25 patients age < 70 and 30 patients age ≥ 70 implanted with the HM II device at a community hospital during the HM II BTT and DT trials. They noted that these patients were carefully selected from 329 total patients referred for consideration. The authors stated, "the main objective of this study was to evaluate the outcomes of LVAD patients ≥ 70 years of age from a community hospital with an experienced LVAD team."
Kaplan-Meier survival estimates were similar for the < 70 and ≥ 70 age groups. The authors did not note any significant differences in the causes of death, adverse event rates, KCCQ, MLHFQ, NYHA classification, 6-MWT, or METS data.
The authors stated, "Selecting the right older patient is critical. Older patients can have more associated illnesses and other concomitant problems with the native heart that need to be considered. Rigorous assessment and optimization of preoperative status should be undertaken, including neurological, nutritional, psychosocial, and renal assessments."
The authors concluded, "Advanced heart failure patients receiving an HM II LVAD who were older than 70 years had outcomes similar to those of patients younger than 70 years. Older patients had acceptable length of hospital stays, adverse events, and functional recovery. Advanced age should not be used as an independent contraindication when selecting a patient for LVAD therapy. As this technology continues to improve, increasing numbers of older patients will seek centers for destination therapy. Analysis of the referral data suggests that more patients should be referred for LVAD evaluation at an experienced center, because good outcomes can be achieved in this patient cohort."
Starling RC, Naka Y, Boyle AJ, Gonzalez-Stawinski G, John R, Jorde U, Russell SD, Conte JV, Aaronson KD, McGee EC Jr, Cotts WG, DeNofrio D, Pham DT, Farrar DJ, Pagani FD. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol. 2011 May 10; 57(19): 1890-8. PMID: 21545946. (Starling, et al., 2011)
Starling et al. reported the results of an FDA-required, manufacturer-sponsored, post-approval study of the HM II for BTT which compared outcomes of the first 169 consecutive patients implanted with the HM II with 169 patients implanted with a pulsatile device. The authors stated, "Post-approval studies were required by the FDA to determine whether results in both trials [BTT and DT] with the device in a commercial setting are comparable to other available devices for the same indication. In the current report we present the findings of the BTT post-approval study in comparison with other FDA-approved ventricular assist devices for BTT."
All patients were enrolled in the INTERMACS registry with a pre-implant strategy of BTT-listed or BTT-likely. Eighty percent of HM II and 89% of pulsatile device patients were receiving intravenous inotropes. Ten percent of HM II and 33% of pulsatile device patients were on an IABP.
Kaplan-Meier survival was 85% at one year for the HM II group (number at risk = 88) and 70% for the pulsatile device group (number at risk = 40).
All adverse event rates were similar or lower in the HM II group.
QOL was measured by the EQ-5D visual analog scale (VAS) with 50% of potential test sessions completed for HM II patients and 44% for pulsatile device patients. QOL improved between pre-implant and three months post-implant and improvements were sustained through one year. Functional capacity was measured by the 6-MWT; however, data were available for fewer than 25% of patients so results were not reported.
The authors concluded, "The results reported demonstrate consistency of outcomes with the HM II LVAD in a post-market approval BTT patient population compared with results from the pivotal clinical trial patient populations. Surprisingly, as the HM II device became available outside of the controlled context of a clinical trial, excellent results were maintained or perhaps surpassed. There is clearly a shift in earlier use of devices in less-ill patients, and currently approximately 35% of patients undergoing heart transplantation in the United States receive some type of mechanical support before transplant. These data suggest that dissemination of this technology has been associated with excellent results and further incremental improvement of outcomes. Importantly, this is the first example of the utility of the INTERMACS registry to conduct a post-market approval study with an LVAD and to demonstrate prospective outcomes with standardized definitions."
John R, Naka Y, Smedira NG, Starling R, Jorde U, Eckman P, Farrar DJ, Pagani FD. Continuous flow left ventricular assist device outcomes in commercial use compared with the prior clinical trial. Ann Thorac Surg. 2011 Oct; 92(4): 1406-13; PMID: 21958789. (John, et al., 2011)
John et al. reported a manufacturer-sponsored retrospective analysis of data from 1,982 HM II BTT recipients: 486 patients implanted during the initial pivotal trial ("trial") and 1,496 implanted after FDA approval and enrolled in the INTERMACS registry ("post-trial"). The authors stated, "The objective of this investigation was to evaluate differences in outcomes for patients supported during the BTT clinical trial and during the post-trial phases over the past 5 years. We believe these results will help clarify the effectiveness of this new technology in real-world clinical use." Ninety percent of trial and 80% of post-trial patients were receiving intravenous inotropes, and 42% and 33% were on an IABP.
Kaplan-Meier survival at one year was 76% in the trial (number at risk = 156) and 85% in the post-trial (number at risk = 393).
Adverse events with similar definitions included bleeding requiring re-exploration (0.23 events/patient-year in the trial vs. 0.12 post-trial), percutaneous lead infection (0.33 vs. 0.28), pump pocket infection (0.03 vs. 0.03), ischemic stroke (0.05 vs. 0.06), hemorrhagic stroke (0.05 vs. 0.02), and device replacement (0.06 vs. 0.02).
QOL was measured using the KCCQ during the trial and the EQ-5D VAS post-trial. KCCQ results were available on 81% of patients pre-implant (393 of 486) and 93% of patients at six months (240 of 258). EQ-5D VAS results were available for 52% of patients pre-implant (777 of 1,496) and 49% at one year (192 of 393). Available data demonstrated improved quality of life in both the trial and post-trial populations. Functional status was measured with the 6-MWT. Data were reported on 100% of patients in the trial population pre-implant and 86% at six months (222 of 258). Data were reported on 72% of patients in the post-trial population pre-implant (1,089 of 1,496) and 57% at six months (469 of 822). Available data demonstrated similar degrees of improvement in both groups.
The authors concluded, "In summary, the results of this study demonstrate that outcomes of patients bridged to transplant with the HeartMate II LVAD have improved since the clinical trial. Results were determined from one of the largest datasets ever evaluated in mechanical circulatory support from a broad range of clinical centers. The findings indicate that excellent outcomes have been maintained with dissemination of new LVAD technology from a clinical trial phase to more broad based use in the postmarket-approval period."
Hsich EM, Naftel DC, Myers SL, Gorodeski EZ, Grady KL, Schmuhl D, Ulisney KL, Young JB. Should women receive left ventricular assist device support?: findings from INTERMACS. Circ Heart Fail. 2012 Mar 1; 5(2): 234-40. PMID: 22260946. (Hsich, et al., 2012)
Hsich et al. compared the outcomes of 401 women and 1,535 men enrolled in the INTERMACS registry. They stated, "Our objective was to determine if there are any sex differences in outcome and adverse events after primary implantation of left ventricular assist devices (LVADs)."
There was no significant difference in mortality or time to first bleed, infection, or device malfunction. However, women had a shorter time to first neurological event that was statistically significant in both unadjusted and adjusted models.
The authors concluded, "In summary, there were no significant sex differences in mortality with either a pulsatile- or a continuous-flow device, but women had a shorter time to first neurological event in both unadjusted and adjusted analyses. Further research is needed to better understand the mechanisms underlying these sex differences."
Kirklin JK, Naftel DC, Kormos RL, Stevenson LW, Pagani FD, Miller MA, Timothy Baldwin J, Young JB. Fifth INTERMACS annual report: risk factor analysis from more than 6,000 mechanical circulatory support patients. J Heart Lung Transplant. 2013 Feb; 32(2): 141-56. PMID: 23352390. (Kirklin, et al., 2013)
Kirklin et al. reported data from 6,561 adult patients receiving an FDA-approved durable VAD through June 30, 2012 at 131 participating facilities. Continuous-flow devices comprised 95% of total and nearly 100% of DT implants.
Actuarial survival for patients with a continuous-flow device was 80% at one year, 70% at two years, and 47% at four years. Risk factors for mortality included older age (hazard ratio (HR) 1.69), INTERMACS profiles 1 (critical cardiogenic shock; HR 2.45) and 2 (progressive decline; HR 1.89), diabetes (HR 1.22), dialysis (HR 2.22), history of cardiac surgery (HR 1.5), and concomitant cardiac surgery (HR 1.34). Over time, fewer patients were implanted in the most severely ill INTERMACS profiles (Table 1).
Table 1: Pre-implant INTERMACS profiles by year.
| INTERMACS Profile |
2006-2007* |
Pre 2011 |
Jan 2011 - Jun 2012 |
| 1 Critical Cardiogenic Shock |
44% |
22% |
16% |
| 2 Progressive Decline |
35% |
42% |
38% |
| 3 Stable but Inotrope dependent |
8% |
19% |
18% |
| 4 through 7 |
12% |
16% |
18% |
* (Kirklin, et al., 2008)
For continuous-flow devices, 41% of patients experienced a major adverse event (infection, bleeding, device malfunction, stroke or death) by one month and 70% by one year. This did not vary significantly between the < 65 and ≥ 65 populations. Actuarial freedom from stroke was 89% at one year and 83% at two years. Thirty-five percent of patients experience a device-related infection (primarily driveline infections) by three years. Freedom from device malfunction leading to device exchange or death was approximately 90% at three years for continuous-flow devices compared with approximately 40% at two years for pulsatile devices.
QOL was measured using the EQ-5D. Data were available for 852 patients pre-implant and 281 patients at 12 months. The authors stated, "Although quality of life data are somewhat limited in MCS [mechanical circulatory support] patients, available data suggest an important and sustained improvement in general well-being, self-care, and usual activities out to at least 1 year."
The authors' bulleted conclusions included, "Greater than 95% of implants are currently continuous- flow devices. Current survival is approximately 80% at 1 year and 70% at 2 years. Elderly patients have generally favorable outcomes but have less tolerance for additional risk factors. Patients in INTERMACS Levels 1 and 2 have about a 5-8% decrease in 1-year survival compared with other INTERMACS levels. Worsening degrees of right ventricular failure and renal dysfunction are associated with an incremental likelihood of early mortality. Adverse event burden will play an important role in driving therapeutic choices for INTERMACS Levels 4 to 7. Quality of life indicators are generally positive after device implant for at least the first year. Major knowledge gaps will be addressed by the addition of dedicated pediatric (PEDIMACS) and medical (MEDAMACS) components within INTERMACS."
INTERMACS Quarterly Statistical Report 2012 4th Quarter http://www.uab.edu/intermacs/images/Federal_Quarterly_Report/INTERMACS_Federal_Partners_Quarterly_Report_03_2012_website_all.pdf (INTERMACS Q4, 2012)
INTERMACS Quarterly Statistical Report Centers for Medicare & Medicaid Services 2012 4th Quarter http:/www.uab.edu/medicine/intermacs/images/CMS/CMS_Report_2012_Q4.pdf (INTERMACS CMS Q4, 2012)
INTERMACS CMS Report Prepared April 21, 2013 http://www.uab.edu/medicine/intermacs/component/content/article/152-uncategorised/161-intermacs-responses (INTERMACS CMS Report, 2013)
INTERMACS investigators reported data from 7,914 adult patients (2,039 age ≥ 65) receiving an FDA-approved durable VAD through December 31, 2012. The registry cannot identify patients who are entitled to Medicare on a basis other than age. During 2011 and 2012, 5,083 patients were implanted and 4,592 patients were enrolled in the registry (90.3%).
In 2012, the majority of patients enrolled were male (80% of total and 85% of age ≥ 65) and white (71% of total and 85% of age ≥ 65). Pre-implant device strategy varied between the total (BTT 19%, BTC 36%, DT 44%) and age ≥ 65 populations (9% BTT, 18% BTC, 72% DT). Fewer older patients were implanted in the most severely ill INTERMACS profiles (Table 2).
Table 2: Pre-implant INTERMACS profiles (2012).
| INTERMACS Profile |
Total |
Age ≥ 65 |
| 1 Critical Cardiogenic Shock |
15% |
10% |
| 2 Progessive Decline |
38% |
35% |
| 3 Stable but Inotrope dependent |
28% |
31% |
| 4 through 7 |
18% |
24% |
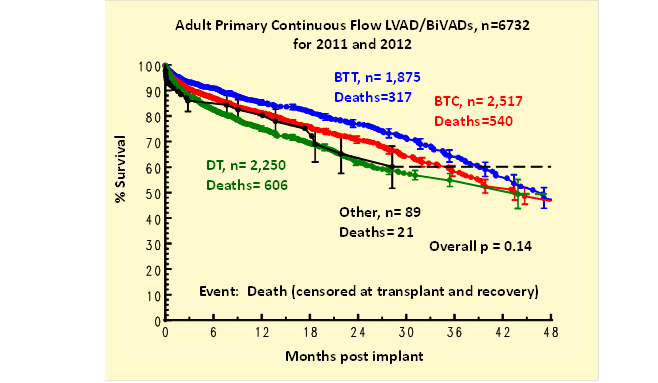
Kaplan-Meier survival (all device types) was less for those age ≥ 65 (Table 3). Survival for BTC patients fell between that of BTT and DT (Figure 1). Adverse events were comparable between the overall population and those age ≥ 65.
Table 3: Kaplan-Meier survival for the total and age ≥ 65 population (Deaths in total population = 1,880; age ≥ 65 = 606).
| Time point |
Total Population
Survival |
Total Population
Number at Risk |
Age ≥ 65
Survival |
Age ≥ 65
Number at Risk |
| 0 Months |
100% |
7,913 |
100% |
2,039 |
| 30-day |
94% |
|
92% |
|
| 6-months |
85% |
4,736 |
81% |
1,240 |
| 1-year |
78% |
2,926 |
72% |
787 |
| 2-year |
66% |
1,150 |
60% |
292 |
| 3-year |
56% |
348 |
47% |
52 |
| 4-year |
47% |
118 |
39% |
13 |
| 5-year |
43% |
|
39% |
|
Figure 1: Kaplan-Meier survival by pre-implant strategy.
Kirklin JK, Naftel DC, Pagani FD, Kormos RL, Stevenson L, Miller M, Young JB. Long-term mechanical circulatory support (destination therapy): on track to compete with heart transplantation? J Thorac Cardiovasc Surg. 2012 Sep; 144(3): 584-603; discussion 597-8. PMID: 22795459. (Kirklin, et al., 2012)
Kirklin et al. reported outcomes of 1,287 adult patients enrolled in the INTERMACS registry with a pre-implant device strategy of DT. They stated "the landscape of devices, their expected durability, and patient outcomes have rapidly evolved over the past 4 years. This study was undertaken to examine, through a national MCS database, the hypothesis that 'mechanical circulatory support as DT has evolved to a level that justifies consideration of selected patients for DT who are transplant eligible.'"
Actuarial survival was 83% at six months, 75% at one year, and 62% at two years. The authors reported that a "hazard ratio of 1.24 reflects the increase in risk from age 60 to 70 years. Perhaps not surprising is the finding that elderly patients receiving DT had a lower general risk profile than did younger patients. Without risk adjustment, the actuarial survival for older and younger patients was similar."
The authors' bulleted conclusions included "DT represents an increasing LVAD application and currently accounts for nearly one third of overall MCS activity in the United States. Evolution from pulsatile to continuous flow technology has dramatically improved 1- and 2-year survivals. DT is not appropriate for patients with rapid hemodynamic deterioration or cardiogenic shock. The presence of severe right ventricular failure is a contraindication for DT. Important subsets of DT patients now enjoy survival that may be competitive with heart transplantation out to about 2 years. Future studies will focus on transplant-eligible subsets who may benefit from informed discussions about MCS as an alternative long-term option."
Brouwers C, Denollet J, de Jonge N, Caliskan K, Kealy J, Pedersen SS. Patient-reported outcomes in left ventricular assist device therapy: a systematic review and recommendations for clinical research and practice. Circ Heart Fail. 2011 Nov; 4(6): 714-23. PMID: 21908585. (Brouwers, et al., 2011)
Brouwers et al. performed a systematic review of the literature on patient-reported outcomes (PROs) in VAD therapy. They concluded, "There is a paucity of studies on the patient perspective of LVAD therapy. Initial evidence suggests an improvement in health status, anxiety, and depression in the first months after LVAD implantation. However, PRO scores of LVAD patients are still lower for physical, social, and emotional functioning compared with transplant recipients. To advance the field of LVAD research and to optimize the management of an increasingly growing population of LVAD patients, more well-designed large-scale studies on PROs are needed. By these studies, we will be able to further elucidate the psychological and social impact of LVAD therapy, thereby creating the opportunity to improve the care for patients after LVAD implantation and to provide important information that is needed by patients and families for effective decision making regarding whether LVAD implantation is aligned with their own preferences and goals."
Question #2: Is the evidence adequate to conclude that maintaining the current facility criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
Slaughter MS, Pagani FD, Rogers JG, Miller LW, Sun B, Russell SD, Starling RC, Chen L, Boyle AJ, Chillcott S, Adamson RM, Blood MS, Camacho MT, Idrissi KA, Petty M, Sobieski M, Wright S, Myers TJ, Farrar DJ; Clinical management of continuous-flow left ventricular assist devices in advanced heart failure. J Heart Lung Transplant. 2010 Apr; 29 (4 Suppl): S1-39. PMID: 20181499. (Slaughter, et al., 2010)
On behalf of the HM II clinical trial investigators, Slaughter et al. "propose key elements in managing patients supported with the new continuous-flow LVADs. Although most of the presented information is largely based on investigator experience during the 1,300-patient HeartMate II clinical trial [for BTT and DT], many of the discussed principles can be applied to other emerging devices as well."
Their observations included:
"A multidisciplinary heart failure team must be organized and charged with providing comprehensive care from initial referral until support is terminated."
"Effective education requires a collaborative, multidisciplinary team approach that extends to the LVAD patient, family member(s), and companion(s) in care."
"Successful long-term LVAD support depends on comprehensive care from a multidisciplinary team, including the patient and his or her family member(s)/caregiver(s)."
"Ongoing follow-up is a key part of effective care for outpatients supported by continuous-flow LVADs. A comprehensive, multidisciplinary team approach to outpatient care may have an important effect on long-term survival."
Adamson RM, Stahovich M, Chillcott S, Baradarian S, Chammas J, Jaski B, Hoagland P, Dembitsky W. Clinical strategies and outcomes in advanced heart failure patients older than 70 years of age receiving the HeartMate II left ventricular assist device: a community hospital experience. J Am Coll Cardiol. 2011 Jun 21; 57(25): 2487-95. PMID: 21679851. (Adamson, et al., 2011)
As discussed under Question #1, Adamson et al. compared outcomes for patients age < 70 with those age ≥ 70 enrolled in the HM II BTT and DT trials. In discussing their results, they stated that "This study demonstrates that destination therapy LVAD therapy can safely be delivered to an older patient population in a small community hospital with an experienced team."
"Optimal outcomes in LVAD patients can be achieved with a dedicated LVAD team organized and charged with implantation, early post-operative management, and outpatient management, as outlined in a recent publication on clinical management of continuous flow LVADs (Slaughter, et al., 2010). Our study shows that if such practices are adopted, then good outcomes can be achieved."
Starling RC, Naka Y, Boyle AJ, Gonzalez-Stawinski G, John R, Jorde U, Russell SD, Conte JV, Aaronson KD, McGee EC Jr, Cotts WG, DeNofrio D, Pham DT, Farrar DJ, Pagani FD. Results of the post-U.S. Food and Drug Administration-approval study with a continuous flow left ventricular assist device as a bridge to heart transplantation: a prospective study using the INTERMACS (Interagency Registry for Mechanically Assisted Circulatory Support). J Am Coll Cardiol. 2011 May 10; 57(19): 1890-8. PMID: 21545946. (Starling, et al., 2011)
As discussed under Question #1, Starling et al. reported the results of the FDA-required post-approval study of the HM II as BTT. In discussing their results, the authors noted that "Heart transplant patient outcomes are related to center volume, cardiologist experience, and dedicated transplant coordinators. Similar observations are emerging that have identified experienced and dedicated patient care teams as critical elements to LVAD patient outcomes."
Pamboukian SV, Tallaj JA, Brown RN, Holman WL, Blood M, George JF, Costanzo MR, Cadeiras M, Smallfield MC, McGiffin DC, Kirklin JK. Improvement in 2-year survival for ventricular assist device patients after implementation of an intensive surveillance protocol. J Heart Lung Transplant. 2011 Aug; 30(8): 879-87. PMID: 21514180. (Pamboukian, et al., 2011)
Pamboukian et al. reported the impact of a disease-management model termed an "intensive surveillance protocol" (ISP) on patient survival following VAD placement. The ISP consisted of consensus-based clinical management practices and three additional components:
- A weekly phone call from a coordinator to the patient or caregiver to identify potential problems if the program had not already been in communication with the patient that week;
- Outpatient care in a multidisciplinary clinic including a cardiologist, cardiovascular surgeon, and nurse coordinator;
- A standardized schedule of clinic visits and protocol of routine diagnostic tests aimed at early detection of complications.
Forty patients were implanted before ISP implementation and 36 after. After risk adjustment, the post-ISP population experienced a 70% reduction in the hazard for death. Adverse event data were not reported.
The authors noted that continuous-flow devices supplanted pulsatile devices during this time so they cannot differentiate the impact of the ISP from the impact of changes in technology. In addition, they could not determine the impact of any temporal changes in patient selection or concurrent management practices over the study period.
The authors concluded, "this study has quantified the benefits of an intensive surveillance protocol in the care of MCSD patients, an approach that was increasingly being practiced in the MCS community before this study, but without clear evidence. We believe this study affirms the current awareness in the MCSD community that optimal outcome after implantation of durable MCSDs requires the same intensive surveillance protocols that are needed for successful heart transplantation. Thus, these patients require an infrastructure of follow-up care that far exceeds that after routine cardiac surgery.
In conclusion, we show that a formalized, long-term management strategy resulted in significant improvements in survival for patients with MCS. After the initial postoperative period, the hazard for death remained constant during the next 24 months. This reinforces the need for long-term vigilance in the management of these patients by clinicians with specialized training and knowledge of the difficulties encountered by this unique patient population."
INTERMACS Quarterly Statistical Report 2012 4th Quarter http://www.uab.edu/intermacs/images/Federal_Quarterly_Report/INTERMACS_Federal_Partners_Quarterly_Report_03_2012_website_all.pdf (INTERMACS Q4, 2012)
INTERMACS Quarterly Statistical Report Centers for Medicare & Medicaid Services 2012 4th Quarter http:/www.uab.edu/medicine/intermacs/images/CMS/CMS_Report_2012_Q4.pdf (INTERMACS CMS Q4, 2012)
INTERMACS CMS Report Prepared April 21, 2013 http://www.uab.edu/medicine/intermacs/component/content/article/152-uncategorised/161-intermacs-responses (INTERMACS CMS Report, 2013)
One hundred and forty-nine were actively enrolled in INTERMACS as of April 2013 compared with 36 facilities in 2006. One hundred and sixteen were credentialed by the Joint Commission for DT and 95 of these were also transplant centers. Thirty-three were not credentialed for DT and 18 of these were also transplant centers.
Hospital implant volume, counting all eligible patients implanted during 2011 and 2012 regardless of whether they were enrolled in INTERMACS, varied significantly (Table 4). This does not include investigational devices which the registry does not track.
Table 4: Quartiles of hospital volume (2011 and 2012).
| Quartile |
Number of Hospitals |
Range of two year implant totals |
|
| 1 |
34 |
1-12 |
| 2 |
35 |
13-32 |
| 3 |
33 |
33-56 |
| 4 |
33 |
57-149 |
|
| Total |
135 |
1-149 |
Kaplan-Meier survival did not vary by hospital volume (Figure 2) or by whether or not a hospital was a heart transplant center (Figure 3).
Figure 2: Kaplan-Meier survival by hospital volume.
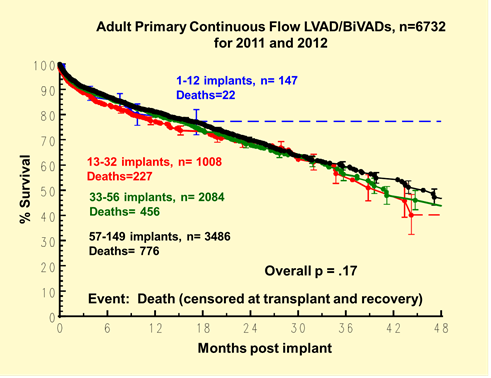
Figure 3: Kaplan-Meier survival by heart transplant center status.
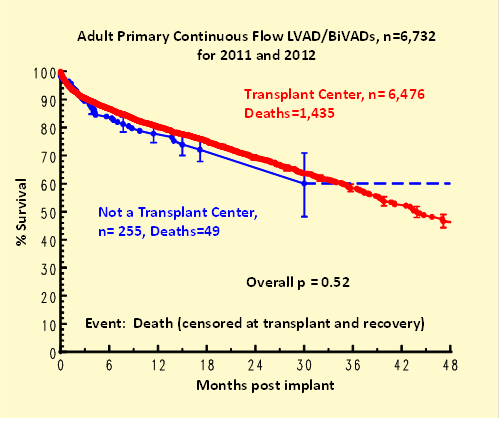
Cowger J, Sundareswaran K, Rogers JG, Park SJ, Pagani FD, Bhat G, Jaski B, Farrar DJ, Slaughter MS. Predicting survival in patients receiving continuous flow left ventricular assist devices: the HeartMate II risk score. J Am Coll Cardiol. 2013 Jan 22; 61(3): 313-21. PMID: 23265328. (Cowger, et al., 2013)
Cowger et al. reported the development of a HM II Risk Score (HMRS) based on data from the HM II BTT and DT trials and CAPs (n = 1,122). The authors stated, "The primary objective of this analysis was to develop and validate a risk model for predicting LVAD candidate outcome in the 'continuous flow era' of MCS. The second objective was to identify predictors of longer-term survival, independent of LVAD operative success."
Key predictors of 90-day mortality following VAD placement included age, albumin, creatinine, INR, and center volume < 15. Center volume was defined as the number of devices implanted by the center during the entire HM II BTT and DT study periods with 15 the threshold at which investigators observed a statistically significant difference in survival. Eighty-eight percent of patients were implanted in a center with a volume of ≥ 15. If patients survived to 90 days postoperative, the only statistically significant preoperative predictors of long-term mortality in the derivation cohort were patient age and center implant volume < 15.
The authors concluded, "Risk factors for mortality after HMII continuous flow LVAD implant in the contemporary LVAD era were identified with a large patient cohort, and a new risk model (the HMRS) was prospectively developed and validated. The HMRS might be useful for patient, family, and referring provider education, providing patient-level LVAD mortality risk assessment regardless of BTT or DT indication. The HMRS also identifies important pre-operative risk factors that might serve as targets for goal-directed interventions meant to improve LVAD candidate survival."
Holman WL, Naftel DC, Eckert CE, Kormos RL, Goldstein DJ, Kirklin JK. Durability of left ventricular assist devices: Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) 2006 to 2011. J Thorac Cardiovasc Surg. 2013 Mar 9. pii: S0022-5223(13) 00168-2. PMID: 23490245. (Holman, et al., 2013)
Holman et al. compared the durability of pulsatile and continuous-flow devices for patients enrolled in the INTERMACS registry. They identified 98 pump exchanges or deaths due to durability issues (3% of implants): 46 in 486 pulsatile device recipients and 52 in 2,816 continuous-flow device recipients. The time to event was significantly greater for continuous-flow devices. Fifty-four percent of continuous-flow and 23% of pulsatile device patients remained alive on device support at one year.
The authors concluded, "The Analysis of Interagency Registry for Mechanically Assisted Circulatory Support data showed greater durability for continuous flow than for pulsatile left ventricular assist devices. Even longer durations of support can be expected if pump durability continues to improve."
4. Medicare Evidence Development & Coverage Advisory Committee (MEDCAC)
CMS convened a MEDCAC meeting on November 14, 2012 to review the evidence for the management of heart failure with the use of VADs.
The panel had intermediate confidence (3.2 out of 5) that there was adequate evidence that specific criteria can be used to prospectively identify patients likely to experience clinically meaningful changes in health outcomes with VAD implant. The chair summarized, "The consistent themes I heard listening to the voting panel were...there is a crying need for more patient-reported outcomes and in particular for really meaningful quality of life and functional status measures, you know, things that would mean something to any of us if we had to make that decision for ourselves or for a loved one..."
The panel had low to intermediate confidence (2.4 of 5) that one or more facility and/or operator characteristics predict clinically meaningful improvements in health outcomes. The chair stated, "To summarize what I heard, and particularly the themes I heard repeated...the heart team, we all agree, is like motherhood and apple pie...perhaps it is because the VAD did grow up in these transplant centers where clearly there was a team...specifying what a team consists of and how important it is in terms of patient care would be really important to outcomes. And certainly when we saw the rapid growth in the VAD centers, it suggested that it is spreading a lot more rapidly."
The panel had intermediate confidence (3.1 of 5) that their conclusions were generalizable to the Medicare beneficiary. The chair summarized, "We made some extrapolations based on age, but it wasn't clear that we were, that particularly since the age of the INTERMACS registry is quite different than the average age of the Medicare population, that that was a reasonable extrapolation. And in addition, all of the things that we're evaluating, quality of life, functional status, adverse events are going to occur at different rates in older people, and the Medicare population in particular have more comorbidities."
Lastly, the panel had high confidence (4.8 of 5) that clinically significant evidentiary gaps remain regarding the use of VADs.
The complete transcript of this meeting can be found at http://www.cms.gov/Regulations-and-Guidance/Guidance/FACA/Downloads/id65c.pdf.
5. Evidence-Based Clinical Guidelines
While several guidelines address the use of VADs, we have focused our review on those based in the United States and the 2013 guideline from the International Society for Heart and Lung Transplantation (ISHLT) which provides the most recent and comprehensive set of recommendations. For each, we outlined the recommendations for facility characteristics and the general recommendations for patient selection but excluded recommendations pertaining to specific comorbidities for brevity. We provide a link to the complete recommendations.
The guidelines label the strength of supporting evidence as follows:
Level A - high-quality randomized controlled trial or high-quality meta-analysis.
Level B - other evidence such as well-designed, nonrandomized clinical trial, lower quality randomized controlled trials, cohort studies, case-controlled studies etc.
Level C - consensus viewpoint or expert opinion.
Feldman D, Pamboukian SV, Teuteberg JJ, Birks E, Lietz K, Moore SA, Morgan JA, Arabia F, Bauman ME, Buchholz HW, Deng M, Dickstein ML, El-Banayosy A, Elliot T, Goldstein DJ, Grady KL, Jones K, Hryniewicz K, John R, Kaan A, Kusne S, Loebe M, Massicotte MP, Moazami N, Mohacsi P, Mooney M, Nelson T, Pagani F, Perry W, Potapov EV, Eduardo Rame J, Russell SD, Sorensen EN, Sun B, Strueber M, Mangi AA, Petty MG, Rogers J. The 2013 International Society for Heart and Lung Transplantation Guidelines for mechanical circulatory support: executive summary. J Heart Lung Transplant. 2013 Feb; 32(2):157-87. PMID: 23352391. (Feldman, et al., 2013)
http://www.ishlt.org/ContentDocuments/JHLT_Feb13_MCS_Guidelines.pdf
The authors presented guidelines for mechanical circulatory support on behalf of the ISHLT. They stated, "that management of patients with MCSDs has been guided by individual clinicians and center-specific protocols. There have been few randomized studies to guide patient selection and care of the MCS patient. Short-term success with MCS therapy largely depends on patient selection, surgical technique, and post-operative management. Long-term success depends on physician and patient engagement in excellent care of their device and personal health."
They noted that, due to limited clinical evidence, most of the recommendations are Level C reflecting expert consensus.
Task Force 1: patient selection for permanent pump implantation including referral and evaluation of potential recipients.
- Long-term MCS for patients who are in acute cardiogenic shock should be reserved for the following (Level of evidence: C):
- Patients whose ventricular function is deemed unrecoverable or unlikely to recover without long-term device support.
- Patients who are deemed too ill to maintain normal hemodynamics and vital organ function with temporary MCSDs, or who cannot be weaned from temporary MCSDs or inotropic support.
- Patients with the capacity for meaningful recovery of end-organ function and quality of life.
- Patients without irreversible end-organ damage.
- Patients who are inotrope-dependent should be considered for MCS because they represent a group with high mortality with ongoing medical management (Level of evidence: B).
- Patients with end-stage systolic heart failure who do not fall into recommendations 1 and 2 above should undergo routine risk stratification at regular intervals to determine the need for and optimal timing of MCS. This determination may be aided by risk assessment calculators and cardiopulmonary stress testing (Level of evidence: C).
- Heart failure patients who are at high-risk for 1-year mortality using prognostic models should be referred for advanced therapy including heart transplant, or MCS (bridge to transplantation [BTT] or destination therapy [DT]) as appropriate (Level of evidence: C).
Task Force 2: patient optimization prior to device implantation.
- A detailed informed consent should discuss the salient aspects of the MCSD placement, common expectations, and possible complications in the peri-operative and post-operative period (Level of evidence: C).
- Palliative care consultation should be a component of the treatment of end-stage heart failure during the evaluation phase for MCS. In addition to symptom management, goals and preferences for end of life should be discussed with patients receiving MCS as DT (Level of evidence: C).
Task Force 3: intraoperative considerations and immediate post-operative care in the intensive care unit.
No recommendations are pertinent to this analysis.
Task Force 4: inpatient management during the post-operative phase, once the patient is out of the intensive care unit through discharge, and during readmission to the hospital.
- A multidisciplinary team led cooperatively by cardiac surgeons and cardiologists and composed of subspecialists (i.e., palliative care, psychiatry, and others as needed), MCS coordinators, and other ancillary specialties (i.e., social worker, psychologist, pharmacist, dietitian, physical therapist, occupational therapist, and rehabilitation services) is indicated for the in-hospital management of MCS patients (Level of evidence: C).
- Health care providers should be trained in MCSD therapy with opportunity to attend refresher classes and ongoing assessment of competency (Level of evidence: C).
- Routine support should be available from social workers, psychologists, or psychiatrists as patients and families adjust to life changes after MCS (Level of evidence: B).
Task Force 5: long-term outpatient care using a multidisciplinary approach.
- Management of the patient with an MCSD should be performed by a multidisciplinary team that includes cardiovascular surgeons, advanced heart failure cardiologists, and specialized MCS coordinators. Other health care providers may collaborate with the primary MCS team when additional expertise is required (Level of evidence: C).
- Between routinely scheduled visits, monitoring phone calls from the MCS coordinator to the patient or caregiver may help proactively identify issues that may adversely affect patient outcomes (Level of evidence: B).
Peura JL, Colvin-Adams M, Francis GS, Grady KL, Hoffman TM, Jessup M, John R, Kiernan MS, Mitchell JE, O'Connell JB, Pagani FD, Petty M, Ravichandran P, Rogers JG, Semigran MJ, Toole JM. Recommendations for the use of mechanical circulatory support: device strategies and patient selection: a scientific statement from the American Heart Association. Circulation. 2012 Nov 27; 126(22): 2648-67. PMID: 23109468. (Peura, et al., 2012) http://circ.ahajournals.org/content/126/22/2648.full.pdf+html
The authors presented recommendations for the use of VADs on behalf of the AHA. They "underscore 2 important principles that have evolved over the past decade. First, some patients are too profoundly ill with multisystem organ failure to benefit from the very best of MCS and aggressive inotropic therapy. Second, complex decisions about candidacy for transplantation or MCS are best made by an experienced, multidisciplinary team. Although it may become appropriate for smaller programs to implant elective DT MCS in highly selected patients, more acutely ill patients should be referred to quaternary care hospitals that are accustomed to the management of such patients."
They stated that there is an "immediate need for more prospective models to the guide the timing of and risk associated with implantation. Major comorbid illness that is anticipated to limit a patient's survival to < 2 years ... should be viewed as a major contraindication to MCS."
Specific recommendations include:
- MCS for BTT indication should be considered for transplant-eligible patients with end-stage HF who are failing optimal medical, surgical, and/or device therapies and at high risk of dying before receiving a heart transplantation [sic] (Level of evidence: B).
- MCS with a durable, implantable device for permanent therapy or DT is beneficial for patients with advanced HF, high 1-year mortality resulting from HF, and the absence of other life-limiting organ dysfunction; who are failing medical, surgical, and/or device therapies; and who are ineligible for heart transplantation (Level of evidence: B).
- Evaluation of potential candidates by a multidisciplinary team is recommended for the selection of patients for MCS (Level of evidence: C).
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013 Jun 5. [Epub ahead of print] PMID: 23741057. (Yancy, et al., 2013) http://circ.ahajournals.org/content/early/2013/06/03/CIR.0b013e31829e8807.long
The authors presented guidelines for care of patients with heart failure on behalf of the American College of Cardiology Foundation (ACCF) and AHA. Specific recommendations include:
- MCS is beneficial in carefully selected patients with stage D HFrEF [heart failure with reduced ejection fraction] in whom definitive management (e.g., cardiac transplantation) or cardiac recovery is anticipated or planned (Level of evidence: B).
- Durable MCS is reasonable to prolong survival for carefully selected patients with stage D HFrEF (Level of evidence: B).
The authors state, "Although optimal patient selection for MCS remains an active area of investigation, general indications for referral for MCS therapy include patients with LVEF < 25% and NYHA class III-IV functional status despite guideline-directed medical therapy, including, when indicated, CRT, with either high predicted 1- to 2-y mortality (e.g., as suggested by markedly reduced peak oxygen consumption, clinical prognostic scores) or dependence on continuous parenteral inotropic support. Patient selection requires a multidisciplinary team of experienced advanced HF and transplantation cardiologists, cardiothoracic surgeons, nurses and, ideally, social workers and palliative care clinicians."
Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010 Jun; 16(6): e1-194. PMID: 20610207. (Lindenfield, et al., 2010)
http://www.heartfailureguideline.org/_assets/document/2010_heart_failure_guideline_exec_summary.pdf
The authors presented guidelines for care of patients with heart failure on behalf of the Heart Failure Society of America (HFSA). Specific recommendations include:
- Patients awaiting heart transplantation who have become refractory to all means of medical circulatory support should be considered for a mechanical support device as a bridge to transplant (Level of evidence: B).
- Permanent mechanical assistance using an implantable assist device may be considered in highly selected patients with severe HF refractory to conventional therapy who are not candidates for heart transplantation, particularly those who cannot be weaned from intravenous inotropic support at an experienced HF center (Level of evidence: B).
Francis GS, Greenberg BH, Hsu DT, Jaski BE, Jessup M, LeWinter MM, Pagani FD, Piña IL, Semigran MJ, Walsh MN, Wiener DH, Yancy CW Jr. ACCF/AHA/ACP/HFSA/ISHLT 2010 Clinical Competence Statement on Management of Patients with Advanced Heart Failure and Cardiac Transplant: A Report of the ACCF/AHA/ACP Task Force on Clinical Competence and Training. J Am Coll Cardiol. 2010 Jul 27; 56(5): 424-53. PMID: 20650365. (Francis, et al., 2010) Circulation. 2010 Aug 10; 122(6): 644-672. PMID: 20644017. (Francis, et al., 2010) http://circ.ahajournals.org/content/122/6/644.full.pdf+html
Francis et al. reported the first statement on clinical competence for the management of patients with advanced heart failure and cardiac transplant endorsed by the ACCF, AHA, American College of Physicians (ACP), HFSA, and ISHLT.
"The purpose of this document is to delineate the knowledge base and skills necessary for expertise in the care and management of patients with advanced heart failure and heart transplantation. It is intended to be used by hospitals, institutions, and credentialing bodies that must at times distinguish specialists in the management of patients with advanced heart failure and heart transplantation from other well-trained physicians who care for the majority of patients with less advanced forms of heart failure."
The recommendations build off of recommendations of the ACCF/AHA/ACP Task Force on Clinical Competence and Training Recommendations reported by Beller et al. which included three tiers of training in cardiology and its subspecialties in ascending levels of intensity. Level 3 heart failure training requires at least one additional year of training in advanced heart failure and transplantation (Beller, et al., 2008).
The authors also addressed institutional competence stating "the committee recognizes that the advanced HF and transplantation specialist should practice in association with an institution that provides the personnel and infrastructure necessary to deliver comprehensive, integrated care. The advanced HF and transplantation specialist should be part of a multidisciplinary team composed of specialists with competence in the management of HF, transplantation, and their associated comorbidities. The availability of cardiothoracic surgeons with expertise in management of high-risk patients with ischemic or valvular heart disease, VADs, and transplantation is essential...The benefits of a comprehensive disease management approach to patients with HF have been well documented in terms of morbidity and long-term survival. The institutional competencies that contribute to successful HF care delivery include the ability to develop and disseminate educational and counseling materials, the inclusion of specialized nurses and/or nurse practitioners, the availability of social service and financial counseling, and the implementation of clinical information systems that facilitate transfer of information among providers. Dietary counseling and cardiac rehabilitation have also been identified as making important contributions to a comprehensive HF management program. The above competencies apply as well to institutions providing care to patients requiring VADs or transplantation, despite the diverse clinical situations associated with these therapies."
6. Professional Society Position Statements
Acker MA, Pagani FD, Stough WG, Mann DL, Jessup M, Kormos R, Slaughter MS, Baldwin T, Stevenson L, Aaronson KD, Miller L, Naftel D, Yancy C, Rogers J, Teuteberg J, Starling RC, Griffith B, Boyce S, Westaby S, Blume E, Wearden P, Higgins R, Mack M. Statement regarding the pre and post market assessment of durable, implantable ventricular assist devices in the United States. J Heart Lung Transplant. 2012 Dec; 31(12): 1241-52. PMID: 23206981. (Acker, et al., 2012) Ann Thorac Surg. 2012 Dec; 94(6): 2147-58. PMID: 23153481. (Acker, et al., 2012) Circ Heart Fail. 2013 Jan; 6(1): 145-50. PMID: 23149496. (Acker, et al., 2013)
Acker et al. summarized a September 16, 2011 meeting of representatives from the FDA, National Institutes of Health National Heart Lung, and Blood Institute (NIH/NHLBI), CMS, medical insurance companies, and industry. Per the authors, "the goal of the meeting was to explore innovative ways to foster the introduction of technologically advanced, safe, and effective ventricular assist devices." They stated, "the summary reflects opinions and conclusions endorsed by The Society of Thoracic Surgeons, American Association for Thoracic Surgery, American Heart Association, Heart Failure Society of America, International Society for Heart and Lung Transplantation, and Interagency Registry of Mechanically Assisted Circulatory Support."
They stated, "From its inception, several features of the INTERMACS registry have distinguished it from traditional registries in terms of scientific rigor, including consistent adverse event definitions, auditing of high priority variables (e.g., terminal events such as death, device exchanges, transplant, or recovery), and a high proportion of patient capture. These features strengthen the quality of the registry, but some limitations remain. Although the majority of patients are entered into the INTERMACS registry, patient inclusion requires informed consent, and some patients decline participation or are missed because of early postoperative death before obtaining informed consent (patients can be enrolled up to 2 weeks following device implant). The INTERMACS results could potentially be skewed if characteristics of patients not entered into the registry differ from those enrolled in the registry. It is also possible that some clinical outcomes may remain undetected, which may bias findings, although audits and data queries are part of the INTERMACS process. Such bias would have important ramifications if the data are used as a comparative control. Outcome data from INTERMACS are likely to be less robust than that generated from a clinical trial where formal clinical endpoint adjudication committees are often used...Finally, because INTERMACS is a real-world registry, standards of care are not controlled as they are in a clinical trial. Thus, different institutions may approach anticoagulation therapy or infection management differently, and these differences have the potential to confound data interpretation...For INTERMACS to serve as a meaningful comparator in future VAD trials, functional assessments need to be collected and recorded at a level commiserate [sic] with a clinical trial."
Participants also discussed whether it was clinically necessary to distinguish between BTT, BTC, and DT indications. The authors stated that "Some view these classifications as clinically artificial, as patients may transition from one designation to another depending on the resolution or occurrence of comorbid conditions or patient choice."
Pagani FD, Kormos RL, Calhoon JH, Higgins RS, Rich JB. Certification for implantation of durable, implantable ventricular assist devices in the United States: the need for clarification of the process. Ann Thorac Surg. 2013 May; 95(5): 1520-2. PMID: 23458402. (Pagani, et al., 2013)
Pagani FD, Acker MA, Camacho MT, Dewey TM, Force SD, McGee EC, McGrath MF, Meyers BF, Mokadam NA, Smedira NG, Toyoda Y, Wallace AF, Weyant MJ. Clinical Statement on the Requirements for Surgeon Certification for Implantation of Durable Ventricular Assist Devices (VADs). Ann Thorac Surg. 2013 May; 95(5): 1834-9. PMID: 23458401. (Pagani, et al., 2013)
Pagani et al. reported consensus-based recommendations for surgeon certification for the implant of VADs on behalf of the Society of Thoracic Surgeons. "The criteria were developed to address a number of important limitations in the interpretation of current CMS requirements for implantation of durable VADs overseen by the Joint Commission."
"The goals of the proposed criteria for surgeon certification are to: 1) establish a minimum standard of training and experience to ensure competency in the technical aspects of VAD implantation and VAD patient management; 2) establish a pathway by which operative and patient care experiences obtained during an ACGME-approved Thoracic Surgery residency or advanced fellowship in cardiac surgery are credited to the CMS volume criterion; 3) establish a pathway by which international or foreign operative and patient care experiences are credited toward the CMS volume criterion; 4) address the unique practice environment of pediatric cardiothoracic surgeons with respect to the volume criterion; 5) address issues of maintenance of VAD certification; and 6) provide clarification of the term "primary surgeon" to provide credit for educational experiences obtained in an ACGME-approved Thoracic Surgery residency, advanced cardiac surgery fellowship or in clinical practice as a board-certified cardiothoracic surgeon."
The full recommendations can be found at http://ats.ctsnetjournals.org/cgi/reprint/95/5/1834.pdf.
7. Public Comments
Initial Public Comment Period - (02/07/2013 - 03/09/2013)
During the initial 30-day public comment period, CMS received 35 public comments addressing patient selection criteria and facility criteria including credentialing, surgeon volume, and hospital staffing requirements. Some commenters favored coverage for BTC patients. Some commenters favored DNV's request to be named as a credentialing organization while others favored the Joint Commission's existing program. One organization questioned the value of any credentialing requirement without stronger evidentiary support.
The comments included fifteen from medical societies and advocacy organizations; one from a health insurance organization; one from a credentialing organization; ten from hospital administrators, medical facilities, physicians and researchers; three from lawmakers; one from a device manufacturer; and four from individuals who did not identify an organization.
The comments can be viewed in their entirety on our website at:
http://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=268
VIII. CMS Analysis
A. Introduction
National coverage determinations are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1862(l) of the Act).
In general, in order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, section 1862(a)(1) of the Social Security Act states, with limited exceptions, no payment may be made under part A or part B for any expenses incurred for items or services:
Which, are not reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member (§1862(a)(1)(A)).
As noted earlier, our review sought answers to the questions below. We have repeated them here for the convenience of the reader.
1. Is the evidence adequate to conclude that maintaining the current patient selection criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
2. Is the evidence adequate to conclude that maintaining the current facility criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
Patient Selection Criteria
Current patient selection criteria for Medicare coverage of durable VADs is based on two broad categories at the time of implant, BTT and DT, which were derived from pivotal VAD trials and FDA-approved device labeling. Medicare has covered VAD implantation as BTT since 1996 and as DT since 2003. Per 2012 INTERMACS data, 19% of devices overall were implanted as BTT and 44% as DT; 9% of devices in patients age ≥ 65 were implanted as BTT and 72% as DT; most of the remainder were implanted as BTC at 36% overall and 18% for patients age ≥ 65 (INTERMACS Q4, 2012) (INTERMACS CMS Q4, 2012). Of note, while these categories are similar to those in our current patient selection criteria, they were not defined specifically to mirror our categories. In addition, the INTERMACS registry does not track insurance status and therefore cannot specifically identify Medicare beneficiaries other than by using age as a proxy for Medicare eligibility.
The MEDCAC and the published literature have questioned both the clinical relevance of characterizing patients pre-implant as BTT, BTC, or DT and the exclusion of the BTC population from Medicare coverage (Felker & Rogers, 2006) (Acker, et al., 2012) (Stevenson & Hunt, 2012) (Miller & Guglin, 2013). We note that the INTERMACS registry includes patients who have been implanted as BTC (labeled in the registry as BTT-likely, BTT-moderately likely, or BTT-unlikely) and that the Kaplan-Meier survival estimates for this category appear similar to that of patients implanted as BTT or DT (INTERMACS CMS Report, 2013). We note, however, that pivotal device trials for FDA approval of VADs have focused separately on BTT and DT without mention of BTC. As a result, CMS coverage has followed sequentially for BTT and DT. We did not identify any trials that have enrolled BTC patients and no devices carry a labeled indication for BTC. Similarly, no trials have enrolled patients independent of these labels. We therefore find the evidence insufficient to justify expansion of coverage to devices implanted as BTC.
We do not currently have specific clinical patient criteria for coverage of VADs intended for use as BTT. The rigorous transplant evaluation and acceptance process ensures a balance of severity of heart failure requiring advanced therapies but limited comorbidity burden that would reduce the chances of a successful transplant. These standards are applicable to VAD placement as well and we note that a primary inclusion criteria for patients enrolled in BTT trials was listing for heart transplant as UNOS status 1A or 1B. We note that our current policy requires a BTT patient to be "listed as a candidate for heart transplantation by a Medicare-approved heart transplant center" but did not specify the referenced list. As we are aware of only one heart transplant waitlist, the national list maintained by the Organ Procurement and Transplantation Network (OPTN), we propose to explicitly state in the NCD that the patient must be active on the OPTN heart transplant waitlist for coverage of a VAD as BTT (HRSA, 2013) (Colvin-Adams, et al., 2012).
Patients who are determined clinically to be ineligible for heart transplant are classified as DT. We currently have several specific patient selection criteria for coverage of VADs as DT which were based on the characteristics of patients enrolled in pivotal trials of continuous-flow devices. The studies reviewed for the current analysis have similar inclusion/exclusion criteria and, as a result, most patients enrolled meet our current criteria. We therefore did not find sufficient evidence to support changes to our patient selection criteria for coverage of devices implanted as DT.
We note that, in fact, pivotal trials have included more stringent exclusions for comorbidities than our current criteria. For instance, patients were excluded for specific comorbidities including severe right ventricular, renal, pulmonary, or hepatic dysfunction, a history of stroke or recent cardiothoracic surgery, or plan for a concomitant procedure at the time of device implant. The INTERMACS registry, however, includes patients with many of these comorbidities and analyses demonstrate that risk factors for morbidity and mortality, not surprisingly, often mirror pivotal trial exclusion criteria (Kirklin, et al., 2013). Similarly, clinical guidelines from the ISHLT and AHA outline relative and absolute contraindications for VAD placement including both specific comorbidities and overall anticipated life expectancy (Feldman, et al., 2013) (Peura, et al., 2012). The AHA notes an "immediate need for more prospective models to the guide the timing of and risk associated with implantation. Major comorbid illness that is anticipated to limit a patient's survival to < 2 years ... should be viewed as a major contraindication to MCS."
Of particular interest to Medicare beneficiaries, several investigators have identified older age as a risk factor for mortality and adverse events; however, others have highlighted that reasonable outcomes can be attained through careful selection of older patients. Adamson et al. found no difference in the outcomes of 25 patients age <70 and 30 patients age ≥ 70 who were consecutively enrolled in HM II BTT and DT studies (Adamson, et al., 2011). Kirklin et al. found similar unadjusted actuarial survival for older and younger patients receiving a VAD as DT, although older patients had a lower general risk profile than younger patients likely reflecting more careful selection (Kirklin, et al., 2012). Mortality and adverse events did not differ between the < 65 and ≥ 65 populations enrolled in the INTERMACS registry (INTERMACS Q4, 2012) (INTERMACS CMS Q4, 2012).
We acknowledge that INTERMACS analyses and clinical guidelines have identified specific pre-operative patient characteristics that significantly increase the risk of morbidity and mortality with VAD implant. However, we also note that many of these same risk factors are likely to increase morbidity and mortality with medical therapy. Since these patients were excluded from randomized trials, we cannot determine the overall balance of risks and benefits of device placement compared with optimal medical therapy for patients with these conditions. We therefore do not propose to incorporate specific comorbid conditions into our patient selection criteria for DT, but will defer to the judgment of experienced clinicians at this time.
Facility Criteria
A. Bridge to Transplant
CMS includes requirements for facility and/or physician standards in certain NCDs where those criteria are needed to ensure that an item or service is reasonable and necessary under the statute. For example, high-risk patients undergoing highly technical procedures such as VAD implantation may require specialized expertise not found in all facilities.
The following facility criteria currently apply to coverage of devices implanted as BTT:
- The implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved heart transplant center under which the patient is listed prior to implantation of the VAD.
- The Medicare-approved heart transplant center should make every reasonable effort to transplant patients on such devices as soon as medically reasonable. Ideally, the Medicare-approved heart transplant centers should determine patient-specific timetables for transplantation, and should not maintain such patients on VADs if suitable hearts become available.
As stated above, we propose that a BTT patient must be active on the OPTN heart transplant waitlist. The transplant center bears ultimate responsibility for the patient being included on the OPTN waitlist, performs the transplant, and eventually tracks the patient's outcomes post-transplant. Therefore, the transplant center is integral to the patient's care and must remain apprised of significant changes in the patient's health status and be involved in treatment decisions including the decision to place a VAD. We reiterate our requirement that "the [VAD] implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved heart transplant center under which the patient is listed prior to implantation."
While we continue to encourage transplant as soon as feasible, our second criterion was established at a time when device durability and longer-term outcomes were unknown. Based on the current evidence, it is no longer necessary to explicitly state this goal and we will remove this criterion for simplicity.
We did not find evidence to support additional changes to our facility criteria for coverage of devices implanted as BTT at this time.
B. Destination Therapy
The following facility criteria currently apply to coverage of devices for DT:
- Facilities must have at least one member of the VAD team with experience implanting at least 10 VADs (as bridge-to-transplant or destination therapy) or artificial hearts over the course of the previous 36 months;
- Facilities must be a member of the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS); and,
- All facilities must have met the above facility criteria and been credentialed by the Joint Commission under the Disease Specific Certification Program for Ventricular Assist Devices (standards dated February 2007).
- Hospitals also must have in place staff and procedures that ensure that prospective VAD recipients receive all information necessary to assist them in giving appropriate informed consent for the procedure so that they and their families are fully aware of the aftercare requirements and potential limitations, as well as benefits, following VAD implantation.
1) Surgeon Requirements
In our review of the evidence, we did not identify any studies to inform modifications to our surgeon volume requirement. We continue to believe that experience is necessary for optimal outcomes of this technically complex procedure and therefore maintain our current requirement. Only surgeons implant VADs and therefore only surgeons qualify as the "member of the VAD team with experience implanting at least 10 VADs;" however, since we outline additional team members below, we propose to state explicitly in this criterion that this individual must be a physician with cardiothoracic surgery privileges. We note that Pagani et al. reported consensus-based recommendations for surgeon certification criteria on behalf of the Society of Thoracic Surgeons which included recommendations for standardized interpretation of this surgeon volume requirement (Pagani, et al., 2013).
2) Dedicated Team Requirements
Several authors have noted the importance of a dedicated VAD team. Slaughter et al. reported observations from the clinical investigators and sites involved in the HM II pivotal studies including a recommendation that "A multidisciplinary heart failure team must be organized and charged with providing comprehensive care from initial referral until support is terminated" and "Successful long-term LVAD support depends on comprehensive care from a multidisciplinary team, including the patient and his or her family member(s)/caregiver(s)." In reporting the results of the FDA-required post-marketing study for the HM II device, Starling et al. stated "observations are emerging that have identified experienced and dedicated patient care teams as critical elements to LVAD patient outcomes" (Starling, et al., 2011). Adamson et al. reported that, for older patients, "optimal outcomes in LVAD patients can be achieved with a dedicated LVAD team organized and charged with implantation, early post-operative management, and outpatient management" (Adamson, et al., 2011). Pamboukian et al. evaluated the impact on survival of an "intensive surveillance protocol (ISP)" consisting of regular calls from a VAD coordinator, outpatient care in a multidisciplinary clinic including a cardiologist, surgeon, and nurse coordinator, and standardized visits and testing (Pamboukian, et al., 2011). Survival was greater following ISP implementation although, due to concurrent improvements in technology and other aspects of clinical care, it was impossible to isolate the impact of the ISP. They note their "study affirms the current awareness in the MCSD community that optimal outcome after implantation of durable MCSDs requires the same intensive surveillance protocols that are needed for successful heart transplantation. Thus, these patients require an infrastructure of follow-up care that far exceeds that after routine cardiac surgery."
Clinical guidelines also highlight the importance of both VAD teams and adequate facility infrastructure. Both the AHA MCS recommendations and the ACCF/AHA heart failure guideline recommend evaluation of potential VAD candidates by a multidisciplinary team (Peura, et al., 2012) (Yancy, et al., 2013). The ACCF/AHA/ACP/HFSA/ISHLT 2010 Clinical Competence Statement on Management of Patients with Advanced Heart Failure and Cardiac Transplant recommends that a heart failure specialist should practice in "an institution that provides personnel and infrastructure necessary to deliver comprehensive, integrated care. The advanced HF and transplantation specialist should be part of a multidisciplinary team composed of specialists with competence in the management of HF, transplantation, and their associated comorbidities" (Francis, et al., 2010). The most recent and comprehensive guideline for VADs, developed by the ISHLT (Feldman, et al., 2013), emphasizes multidisciplinary team-based care including the following:
- For in-hospital management, they recommend care by a multidisciplinary team led cooperatively by cardiac surgeons and cardiologists and composed of subspecialists (i.e., palliative care, psychiatry, and others as needed), MCS coordinators, and other ancillary specialties (i.e., social worker, psychologist, pharmacist, dietitian, physical therapist, occupational therapist, and rehabilitation services).
- For outpatient management, they recommend care by a multidisciplinary team that includes cardiovascular surgeons, advanced heart failure cardiologists, and specialized MCS coordinators. Other health care providers may collaborate with the primary MCS team when additional expertise is required.
- Palliative care consultation should be a component of the treatment of end-stage heart failure during the evaluation phase for MCS.
- Routine support should be available from social workers, psychologists, or psychiatrists as patients and families adjust to life changes after MCS.
- Health care providers should be trained in MCSD therapy with opportunity to attend refresher classes and ongoing assessment of competency.
- A detailed informed consent should discuss the salient aspects of the MCSD placement, common expectations, and possible complications in the peri-operative and post-operative period.
Based on these recommendations, we propose to require that patients receiving VADs for DT be cared for by a team defined as a cohesive, multi-disciplinary team of medical professionals with the appropriate qualifications, training, and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in shared decision making and to provide appropriate informed consent.
In addition to the cardiothoracic surgeon noted above, we propose that the team must have, at a minimum, one cardiologist trained in advanced heart failure with clinical competence in medical and device-based management including VADs and clinical competence in the management of patients before and after heart transplant, a VAD coordinator, a social worker, and a palliative care specialist. All team members must be based at the facility and have experience with patients before and after placement of VADs. Our previous requirement for informed consent is now encompassed in the definition of the VAD team so we propose to remove this separate statement to avoid duplication.
3) Hospital Requirements
We identified two studies evaluating the impact of hospital characteristics on the outcomes of patients receiving durable, continuous-flow devices. Cowger et al. found that hospital experience was a significant predictor of 90-day mortality following VAD implant (OR 2.24, 95% confidence interval 1.15 to 4.37) (Cowger, et al., 2013). In their model, experience was dichotomized at 15 implants over the course of the entire HM II BTT and DT study and CAP periods (2005-2010). In the second study, INTERMACS investigators did not find differences in survival between quartiles of total hospital implant volume in unadjusted models (INTERMACS CMS Report, 2013). While the Cowger study might support a hospital-level implant volume requirement, it is based on initial device experience at highly selected pivotal trial centers summed over several years. The generalizability is uncertain. In contrast, INTERMACS analyses did not identify a relationship with hospital volume.
When we initially covered VADs for DT in 2003, we stated that "CMS has significant concerns as to whether the results obtained in clinical trials of new and high-risk procedures can be matched in all Medicare-approved facilities" and that "Because the length of survival to be expected for destination therapy is still unknown, CMS believes that long term outcomes data on all recipients is needed to provide patients with the information that they need in making an informed choice about whether to have this surgery. Thus, CMS concludes that collection of data by a registry is crucial to provide access to information necessary to support advancement of this technology." We further stated that VADs represented "a complex device requiring a technically demanding surgical procedure for proper implantation. The procedure, the device and post-operative care will continue to be refined in coming years and it is important to have a means of assessing the quality of patient care over time to ensure that outcomes are maintained or ideally improved. Registry data will permit facilities to compare their LVAD experience against that of other implanting facilities to determine the quality of their performance overall as well as to assess whether an individual patient's care and progress in recovering from the procedure is meeting normative standards... Registry data can be an invaluable aid to an implanting facility in ongoing assessment of the quality of care it is providing to its patients."
For these reasons, we have required registry participation since 2003 and named INTERMACS as the required registry in 2007. This requirement was intended to fulfill three primary goals discussed above: (1) to provide information on longer-term outcomes for DT, (2) to evaluate whether outcomes were maintained beyond the carefully selected trial sites, and (3) to inform program quality assessment and improvement (e.g., refinement of the procedure and post-operative care, ongoing assessment of quality of care, comparing facility experience against that of others).
We have sufficient data to answer the first question posed by our registry requirement. Our 2003 decision was based primarily on the Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure trial (REMATCH) which randomized patients ineligible for heart transplant to placement of the pulsatile HM VE or optimal medical management (Rose, et al., 2001). Survival of patients assigned to the HM VE was 52% at one year compared with 25% for medical therapy alone. Since that time, both survival and adverse event rates have improved, reportedly due to refinements in patient selection, improvements in medical management, and transition to continuous-flow technology (Appendix B: Table 5, Table 6). INTERMACS analyses confirm these findings with survival of 75% at one year, 62% at two years, and approximately 50% at four years (Kirklin, et al., 2012) (INTERMACS CMS Report, 2013).
We also have sufficient data to answer the second question posed by our registry requirement. We were initially concerned that outcomes might not be generalizable beyond the small number of carefully selected heart transplant centers that participated in the pivotal trials. Since that time, the number of facilities implanting VADs has grown from 36 in 2006 to 149 in 2012 (INTERMACS Q4, 2012). While we initially restricted DT implants to Medicare-certified heart transplant centers, we removed this requirement in 2007 and 36 of the 149 hospitals currently enrolled in INTERMACS do not perform heart transplants. Despite significant expansion to non-study sites as well as to non-transplant centers, post-marketing studies of the HM II device and INTERMACS analyses have demonstrated similar or improved survival and lower adverse event rates compared with the initial trials (Appendix B: Table 5, Table 6) (Starling, et al., 2011) (John, et al., 2011) (Kirklin, et al., 2013) (INTERMACS CMS Report, 2013).
We note progress on the third goal as evidenced by data-driven refinements in care practices such as anticoagulation and infection management as well as a trend toward fewer implants in the most critically ill patients who are known to be higher risk. These improvements have resulted in improvements in both mortality and serious adverse events such as bleeding, stroke, and device-related infection (Appendix B: Table 5, Table 6).
To summarize, in the ten years since we first required registry participation, we have observed tremendous growth in the number of hospitals implanting VADs and the number of patients implanted, and we have followed patients for longer periods of time. Despite this rapid expansion, we have observed improvements in survival with fewer adverse events. The current data are adequate to conclude that VADs are reasonable and necessary for Medicare beneficiaries who meet our current selection criteria. We therefore propose to remove the requirement that facilities implanting devices as DT be a member of the INTERMACS registry.
We acknowledge that the third goal of quality improvement is perpetual for any medical intervention but particularly so for VAD implantation, which remains a high-risk procedure with significant morbidity and mortality. We therefore believe that it is important for facilities to continue to track their outcomes in a way that allows comparisons with other institutions to facilitate internal monitoring and quality improvement and to further refine clinical practices. We propose to include a requirement that facilities track patient outcomes including survival, adverse events (e.g. bleeding, infection, stroke, and device malfunction), functional status, and quality of life in a way that allows comparisons with other institutions and facilitates internal quality monitoring and improvement.
Since 2007, we have required credentialing by the Joint Commission under the Disease Specific Certification Program for Ventricular Assist Devices (standards dated February 2007) for facilities implanting VADs as DT. We believed this was necessary for several reasons that remain relevant: advanced heart failure patients and VAD recipients are medically fragile; the procedure is high risk and technically complex; this is a time of rapid dissemination with continued growth in both the number of hospitals implanting VADs and the number of patients implanted, especially as DT; and the community is just beginning to establish consensus-based standards. While we did not identify any studies assessing the impact of facility credentialing on patient outcomes, for the reasons outlined, we will continue to require credentialing of facilities seeking to implant VADs as DT at this time.
At the time of our initial requirement, the Joint Commission program was the only VAD-specific credentialing program. The external requestor for this reconsideration asks to be included in this NCD as an acceptable credentialing organization. We find insufficient reason to continue to restrict credentialing to one organization. Therefore, we propose that credentialing organizations with standards that ensure that facilities meet the criteria outlined in the final NCD are eligible to be recognized as a Medicare-approved credentialing organization for purposes of this decision. These organizations may include additional requirements necessary to operationalize these criteria or administer the credentialing program. Additional substantive standards not outlined in the final NCD are not required by the NCD for Medicare coverage.
As of July 2013, 127 facilities were credentialed by the Joint Commission and listed on the CMS website as Medicare-approved facilities for VADs as DT. We do not believe it would be practical to require these credentialed facilities to undergo immediate re-credentialing to verify compliance with these updated criteria. Patients who are candidates for a VAD as DT have no other life-saving therapeutic options. The clinical need for a VAD cannot always be predicted in advance or delayed once a patient's condition worsens; therefore, it is in the public interest to ensure a seamless transition that maintains continuous access to this procedure for Medicare beneficiaries. Therefore we propose a one year transition period to allow facilities currently credentialed by the Joint Commission to adjust their staffing and clinical operations to meet the new facility criteria and to allow credentialing organizations to update their program standards, request and obtain CMS approval to credential facilities to implant VADs as DT, and to verify compliance of existing facilities with the new criteria. Therefore, we propose that facilities currently credentialed by the Joint Commission for placement of VADs as DT will remain on the list of Medicare-approved facilities for one year following posting of the final decision to allow for a seamless transition to the new facility criteria. Facilities that have not demonstrated to their credentialing organization that they are in compliance with the new criteria by the end of this period will be removed from the list of approved facilities.
As we reconsidered the credentialing requirement for facilities implanting VADs as DT, we reviewed several options. We considered approving additional credentialing organizations but keeping the list of approved organizations within the NCD itself; however, this would require a reconsideration of the NCD each time we needed to update the list which would limit our ability to respond quickly to changes that would enhance protections for our beneficiaries or efficiencies for facilities and practitioners. We also considered eliminating the credentialing requirement altogether with or without requiring facilities to attest to meeting the facility criteria. At this time we do not believe removing credentialing is adequate for this highly technical and high-risk procedure during a time of continued rapid evolution and growth.
Limitations of the Current Evidence
Since our last NCA, the number of patients implanted with durable VADs has grown substantially and the number of implanting facilities has expanded well beyond the initial clinical trial sites. Destination therapy, the most recent FDA-approved indication, now accounts for over 70% of VAD implants in patients age 65 and over. Despite the growth in this field, we identified significant limitations in the literature for the use of durable VADs which impact our ability to refine patient selection and facility criteria.
Well-designed, double-blind, randomized controlled trials provide the highest level of evidence as subjects are allocated to comparison groups in an unbiased way and outcomes are assessed without knowledge of the assigned treatment. In the absence of new randomized trials, we reviewed non-randomized trials which introduced the potential for several important biases.
First, the population studied may differ significantly from the comparison group making it challenging to discern the impact of the VAD implant itself on patient outcomes. This is an important consideration for studies of VADs as evidenced, for example, by decreasing proportions of patients dependent on intravenous inotropes or an IABP over time (Appendix B: Table 5, Table 6). In addition, fewer patients are being implanted in the most severe INTERMACS profiles. For instance, 44% of patients were implanted with pre-implant INTERMACS profile 1 (critical cardiogenic shock) between June 2006 and December 2007 compared with only 16% between January 2011 and June 2012 (Kirklin, et al., 2008) (Kirklin, et al., 2013).
Several of the studies we reviewed were uncontrolled. In an uncontrolled trial, it can be difficult to differentiate the impact of the device from that of other heart failure therapies, especially as medical management continues to improve over time. This makes it challenging to identify the point at which the benefits of VAD implant exceed the benefits of current medical management. We note that The Evaluation of VAD InterVEntion Before Inotropic Therapy (REVIVE-IT) trial will randomize patients with a lesser illness severity to the HM II device or continued optimal medical therapy in an attempt to address some of these questions (ClinicalTrials.gov Identifier NCT01369407) (Baldwin & Mann, 2010). In addition, we note the launch of the MedaMACS registry which will study medically managed advanced heart failure patients (UAB School of Medicine, 2013).
Blinding is generally not feasible for studies of VADs due to differences in surgical implantation, device function, and external hardware. In an unblinded study, ascertainment of outcomes may be biased especially for those outcomes with a subjective component or those that reflect device-related harms. This is particularly true in a registry setting where adverse events and outcomes are not easily audited and cannot achieve the level of oversight provided for clinical trials. For instance, in the INTERMACS registry, even death and heart transplant cannot be confirmed with external sources such as the Social Security Death Index or the OPTN database since patient identifiers are not captured. Similarly, during 2011 and 2012, INTERMACS hospitals enrolled 90.3% of patients receiving FDA-approved durable VADs. Missing patients may bias outcomes if they differ in significant ways from those enrolled such as failing to consent to registry participation due to early mortality.
As VADs are considered for less severely ill patients and for longer durations of support, it is even more critical to consider the patient's experience including their physical function and quality of life. Morbidity is significant with 41% of patients experiencing a major adverse event (infection, bleeding, device malfunction, stroke, or death) by one month and 70% by one year (Kirklin, et al., 2013). Understanding the overall impact of VAD therapy, as experienced by patients themselves, will more clearly portray expectations and enhance patients' ability to make informed decisions. Unfortunately, our review identified significant limitations to existing functional status and quality of life assessments. We found substantial missing data and cannot exclude the possibility that patients completing assessments differ in important ways from those that do not (e.g. illness severity, burden of adverse events). For BTT populations, it is also possible that patients experiencing worse outcomes are preferentially "rescued" by heart transplant, thereby leaving patients with more favorable experiences on device therapy. Although the INTERMACS registry is able to collect these data, the results are uninterpretable due to severe underreporting. Lastly, patient-reported outcomes can be particularly prone to bias in an unblinded study, as patients who enroll in a clinical trial may have preconceived beliefs that they will benefit from the intervention.
We note that all of the trials and several of the INTERMACS registry analyses we identified were sponsored by device manufacturers which may result in a more positive portrayal of the device in the literature if favorable findings are preferentially explored and published.
Finally, in this and previous analyses we note that we have not reviewed the evidence for the use of VADs for right ventricular support either alone or in combination with a left-sided device for biventricular support (implanted either simultaneously or sequentially). We also have not reviewed the evidence for VAD implantation for pediatric indications or for adults with complex congenital heart disease. As such, we propose to add a statement that the NCD does not address coverage of VADs for right ventricular support, biventricular support, use in patients under the age of 18, or use in patients with complex congenital heart disease and that coverage for items and services under section 1862(a)(1)(A) in these situations will be made by local Medicare Administrative Contractors (MACs) within their respective jurisdictions.
Summary
In summary, we believe the answer is yes for question one: "Is the evidence adequate to conclude that maintaining the current patient selection criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?"
For coverage of VADs as BTT, we propose that the patient must be active on the OPTN heart transplant waitlist as we are not aware of any other official list. We maintain our requirement that the implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved heart transplant center under which the patient is listed prior to implantation of the VAD. Lastly, we find the evidence sufficient to remove the current BTT requirement that a "Medicare-approved heart transplant center should make every reasonable effort to transplant patients on such devices as soon as medically reasonable. Ideally, the Medicare-approved heart transplant centers should determine patient-specific timetables for transplantation, and should not maintain such patients on VADs if suitable hearts become available."
For coverage of VADs as DT, we find the evidence insufficient to support changes to our patient selection criteria. We therefore maintain our current patient selection criteria as follows:
The VADs are covered for patients who have chronic end-stage heart failure (New York Heart Association Class IV end-stage left ventricular failure) who are not candidates for heart transplantation, and meet all of the following conditions:
- Have failed to respond to optimal medical management (including beta-blockers and ACE inhibitors if tolerated) for at least 45 of the last 60 days, or have been balloon pump-dependent for 7 days, or IV inotrope-dependent for 14 days; and,
- Have a left ventricular ejection fraction (LVEF) < 25%, and,
- Have demonstrated functional limitation with a peak oxygen consumption of < 14 ml/kg/min unless balloon pump- or inotrope-dependent or physically unable to perform the test.
We find the evidence insufficient to justify coverage of devices implanted as BTC.
In summary, we found the answer to vary by criterion for question two: Is the evidence adequate to conclude that maintaining the current facility criteria for VAD implantation will provide improved health outcomes for Medicare beneficiaries?
We found the evidence sufficient to conclude that facilities that meet certain criteria provide improved health outcomes for Medicare beneficiaries. We are proposing to update our facility criteria as a condition of coverage of this procedure as DT under section 1862(a)(1)(A). At a minimum, these facilities must meet the following criteria:
- Patients receiving VADs for DT must be cared for by a team defined as a cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in shared decision making and to provide appropriate informed consent.
The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum, all of the following:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 VADs over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical and device-based management including VADs, and clinical competence in the management of patients before and after heart transplant.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
- Facilities must track patient outcomes including survival, adverse events (e.g. bleeding, infection, stroke, and device malfunction), functional status, and quality of life in a way that allows comparisons with other institutions and facilitates internal quality monitoring and improvement.
- Facilities must be credentialed by an organization approved by CMS.
We found the evidence sufficient to conclude that continuing required participation in the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) will not adequately address the outstanding evidentiary questions for VADs; therefore, INTERMACS participation is no longer required for VADs to be determined reasonable and necessary and we propose to remove this requirement.
We maintain our requirement that VADs used as BTT or DT are covered only if they have received approval from the FDA for that purpose, and the VADs are used according to the FDA-approved labeling instructions.
B. Disparities
Women comprise more than half of all patients diagnosed with heart failure in the United States but only 20% of patients enrolled in VADs trials and 20% of commercial implants (15% of patients age ≥ 65) (Adams, et al., 2005) (INTERMACS CMS Q4, 2012) (INTERMACS CMS Q4, 2012). Bogaev et al. demonstrated similar survival in men and women enrolled in the HM II BTT trial and CAP but women experienced a higher rate of hemorrhagic stroke (0.10 events/patient-year vs. 0.04) and a lower rate of device-related infections (0.23 vs. 0.44) (Bogaev, et al., 2011). Hsich et al. compared outcomes for men and women enrolled in the INTERMACS registry and found no difference in mortality or most adverse events; however, women had a shorter time to first neurological event (Hsich, et al., 2012). The significant disparity between the number of women with heart failure and those reflected in the INTERMACS registry as VAD recipients has been attributed in the past, at least in part, to anatomic constraints as some women were unable to accommodate larger pulsatile pumps. This should be monitored with the diffusion of smaller devices.
Blacks have a higher prevalence of heart failure compared with whites and often present at a younger age and more advanced stage (Bahrami, et al., 2008) (Heidenreich, et al., 2013). They comprise between 15 and 30% of clinical trial populations although race is often not reported. They account for 21% of the total INTERMACS population, but only 9% of those age ≥ 65 (INTERMACS Q4, 2012) (INTERMACS CMS Q4, 2012). These differences warrant further investigation.
IX. Conclusion
The Centers for Medicare & Medicaid Services (CMS) proposes the following changes to the current national coverage determination (NCD).
- Ventricular assist devices (VADs) for bridge to transplant (BTT)
- For the existing requirement that a patient is approved and listed as a candidate for heart transplant by a Medicare-approved heart transplant center, we clearly identify that the patient must be active on the waitlist maintained by the Organ Procurement and Transplantation Network.
- Remove the existing requirement that a "Medicare-approved heart transplant center should make every reasonable effort to transplant patients on such devices as soon as medically reasonable. Ideally, the Medicare-approved heart transplant centers should determine patient-specific timetables for transplantation, and should not maintain such patients on VADs if suitable hearts become available."
- VADs for destination therapy (DT)
- The evidence is insufficient to support changes to our current patient selection criteria for coverage of a VAD as DT.
- The evidence is sufficient to conclude that VADs implanted in facilities that meet certain criteria improve health outcomes for Medicare beneficiaries. Facilities currently credentialed by the Joint Commission for placement of VADs as DT may continue as Medicare-approved facilities for a period of one year following the posting of the final decision memorandum. At the conclusion of this transition period, these facilities must be in compliance with the following criteria as determined by the credentialing organization. As of the effective date, new facilities, must meet the following criteria as a condition of coverage of this procedure as DT under section 1862(a)(1)(A):
-
Beneficiaries receiving VADs for DT must be managed by an explicitly identified cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in shared decision making and to provide appropriate informed consent.
The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum, all of the following:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 VADs over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical and device-based management including VADs, and clinical competence in the management of patients before and after heart transplant.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
- Facilities must track patient outcomes including survival, adverse events (e.g. bleeding, infection, stroke, and device malfunction), functional status, and quality of life in a way that allows comparisons with other institutions and facilitates internal quality monitoring and improvement.
- Facilities must be credentialed by an organization approved by CMS.
We propose to remove the separate requirement that hospitals have in place staff and procedures for appropriate informed consent as this requirement is encompassed in the above team definition.
The evidence is sufficient to conclude that continuing required participation in the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) will not adequately address the outstanding evidentiary questions for VADs; therefore, INTERMACS participation is no longer required for VADs to be determined reasonable and necessary and CMS proposes to remove this requirement.
We propose to allow organizations that have credentialing programs specific to VADs to apply to CMS to be designated as a credentialing organization for VAD facilities for DT. These programs must ensure that credentialed facilities meet the criteria outlined in the NCD and may include additional requirements necessary to operationalize these criteria or administer the credentialing program. Beyond this, any additional substantive credentialing standards not identified in the final NCD are not required for Medicare coverage.
The process for organizations to apply for CMS approval to be designated as a credentialing organization for VAD facilities for DT will be posted on our web site along with a list of approved credentialing organizations, approved standard versions, and credentialed facilities. http://www.cms.gov/Medicare/Medicare-General-Information/MedicareApprovedFacilitie/VAD-Destination-Therapy-Facilities-Aug2007.html.
We propose to add a statement that the NCD does not address coverage of VADs for right ventricular support, biventricular support, use in patients under the age of 18, or use in patients with complex congenital heart disease and that coverage for items and services under section 1862(a)(1)(A) in these situations will be made by local Medicare Administrative Contractors (MACs) within their respective jurisdictions.
CMS is seeking comments on our proposed decision. We will respond to public comments in a final decision memorandum, as required by §1862(l)(3) of the Social Security Act (the Act).
In addition to the proposed changes above, CMS is renumbering its VAD-related policies into a sub-section of section 20.9 (Artificial Hearts and Related Devices) of the NCD Manual. The sub-section (20.9.1) will be titled Ventricular Assist Devices. This is an administrative change only to make it easier for the public to read and understand the VAD policies. Section 20.9.1 will include the existing coverage of VADs for postcardiotomy, BTT, and DT.
The changes to the manual are reflected in Appendix C.
Appendix A: General Methodological Principles of Study Design
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of an illness or injury or to improve the functioning of a malformed body member. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention's potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to help ensure adequate numbers of patients are enrolled to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to which group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is the extent to which differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well-designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of which have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
- Randomized controlled trials
- Non-randomized controlled trials
- Prospective cohort studies
- Retrospective case control studies
- Cross-sectional studies
- Surveillance studies (e.g., using registries or surveys)
- Consecutive case series
- Single case reports
When there are merely associations but not causal relationships between a study's variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in which confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to which the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study's external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator's lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention's potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study's selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. We are interested in the results of changed patient management not just altered management. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention's benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. For most determinations, CMS evaluates whether reported benefits translate into improved health outcomes. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology's benefits and risk of harm to Medicare beneficiaries.
Appendix B: Tables
Table 1: Survival in destination therapy studies
| |
Study design |
Device |
Population |
#Patients/
Centers |
%inotrope
dependent/
%IABP |
12-month
Survival |
2-year
Survival |
Rose
2001 |
Randomized controlled trial
Pivotal study |
Medical
Management |
NYHA class
IV |
61/20 |
72/ |
25% |
8% |
| |
|
HM VE |
NYHA class
IV |
68/20 |
65/ |
52% |
8% |
Slaughter
2009 |
Randomized controlled trial
Pivotal study |
HM XVE |
NYHA class
IIIB or IV |
66/38 |
83/23 |
55% |
24% |
| |
|
HM II |
NYHA class
IIIB or IV |
134/38 |
77/22 |
68% |
58% |
Park
2012 |
Follow-up of HM II DT trial and CAP |
HM II |
NYHA class
IIIB or IV |
281/38 |
78/19 |
73% |
63% |
Kirkin
2012 |
Retrospective analysis of INTERMACS registry data |
90% HM II |
Enrolled in INTERMACS as DT |
1,287/104 |
/ |
75% |
62% |
Table 2: Adverse events in destination therapy studies for continuous flow devices (events/patient-year).
| |
Population |
Bleeding
Requiring
Transfusion |
Bleeding
Requiring
Surgery |
Sepsis |
Device-related
Infection |
Ischemic
Stroke |
Hemorrhagic
Stroke |
Device
Replacement |
Slaughter
2009 |
HM XVE |
2.45 |
0.29 |
1.11 |
0.90 |
0.10 |
0.12 |
0.51 |
Slaughter
2009 |
HM II |
1.66 |
0.23 |
0.39 |
0.48 |
0.06 |
0.07 |
0.06 |
| Park 2012 |
HM II CAP |
1.13 |
0.14 |
0.27 |
0.27 |
0.05 |
0.03 |
0.04 |
Appendix C: Draft of Section 20.9 Artificial Hearts and Related Devices and Section 20.9.1 Ventricular Assist Devices
20.9 - Artificial Hearts and Related Devices (Various Effective Dates Below)
(Rev.)
A. General
An artificial heart is a biventricular replacement device which requires removal of a substantial part of the native heart, including both ventricles. Removal of this device is not compatible with life, unless the patient has a heart transplant.
B. Nationally Covered Indications
1. Bridge-to-transplant (effective for services performed on or after May 1, 2008)
An artificial heart for bridge-to-transplantation is covered when performed under coverage with evidence development (CED) when a clinical study meets all of the criteria listed below. The clinical study must address at least one of the following questions:
- Were there unique circumstances such as expertise available in a particular facility or an unusual combination of conditions in particular patients that affected their outcomes?
- What will be the average time to device failure when the device is made available to larger numbers of patients?
- Do results adequately give a reasonable indication of the full range of outcomes (both positive and negative) that might be expected from more widespread use?
The clinical study must meet all of the criteria stated in Section D of this policy.
The above information should be mailed to: Director, Coverage and Analysis Group, Centers for Medicare & Medicaid Services (CMS), Re: Artificial Heart, Mailstop S3-02-01, 7500 Security Blvd, Baltimore, MD 21244-1850.
Clinical studies that are determined by CMS to meet the above requirements will be listed on the CMS Web site at: http://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/Artificial-Hearts.html.
2. Destination therapy (effective for services performed on or after May 1, 2008)
An artificial heart for destination therapy is covered when performed under CED when a clinical study meets all of the criteria listed below. The clinical study must address at least one of the following questions:
- Were there unique circumstances such as expertise available in a particular facility or an unusual combination of conditions in particular patients that affected their outcomes?
- What will be the average time to device failure when the device is made available to larger numbers of patients?
- Do results adequately give a reasonable indication of the full range of outcomes (both positive and negative) that might be expected from more widespread use?
The clinical study must meet all of the criteria stated in Section D of this policy.
The above information should be mailed to: Director, Coverage and Analysis Group, Centers for Medicare & Medicaid Services, Re: Artificial Heart, Mailstop S3-02-01, 7500 Security Blvd, Baltimore, MD 21244-1850.
Clinical studies that are determined by CMS to meet the above requirements will be listed on the CMS Web site at: http://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/Artificial-Hearts.html.
C. Nationally Non-Covered Indications
All other indications for the use of artificial hearts not otherwise listed remain non-covered, except in the context of Category B investigational device exemption clinical trials (42 CFR 405) or as a routine cost in clinical trials defined under section 310.1 of the National Coverage Determinations (NCD) Manual.
D. Other
Clinical study criteria:
- The study must be reviewed and approved by the Food and Drug Administration (FDA).
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants' health outcomes.
- The research study is well supported by available scientific and medical information, or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found at 45 CFR Part 46. If a study is FDA-regulated it also must be in compliance with 21 CFR Parts 50 and 56.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed here as Medicare requirements for coverage with evidence development (CED).
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the ClinicalTrials.gov Web site by the principal sponsor/investigator as demonstrated by having a ClinicalTrials.gov Identifier.
- The research study protocol specifies the method and timing of public release of all pre-specified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer-reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (ICMJE) (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally under-represented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of under-represented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability, or Medicaid eligibility.
Consistent with section 1142 of the Social Security Act (the Act), the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
The principal investigator of an artificial heart clinical study seeking Medicare payment should submit the following documentation to CMS and should expect to be notified when the CMS review is complete:
- Complete study protocol (must be dated or identified with a version number);
- Protocol summary;
- Statement that the submitted protocol version has been agreed upon by the FDA;
- Statement that the above study standards are met;
- Statement that the study addresses at least one of the above questions related to artificial hearts;
- Complete contact information (phone number, email address, and mailing address); and,
- Clinicaltrials.gov Identifier.
(This NCD last reviewed XXX.)
20.9.1 - Ventricular Assist Devices (Various Effective Dates Below)
A. General
A ventricular assist device (VAD) is a surgically implanted device intended to assist or augment the ability of a damaged or weakened native heart to pump blood. Improvement in the performance of the native heart may allow the device to be removed.
B. Nationally Covered Indications
1. Postcardiotomy (effective for services performed on or after October 18, 1993)
Post-cardiotomy is the period following open-heart surgery. VADs used for support of blood circulation post-cardiotomy are covered only if they have received approval from the Food and Drug Administration (FDA) for that purpose, and the VADs are used according to the FDA-approved labeling instructions.
2. Bridge to Transplant (effective for services performed on or after January 22, 1996)
The VADs used for bridge to transplant are covered only if they have received approval from the FDA for that purpose, and the VADs are used according to FDA-approved labeling instructions. All of the following criteria must be fulfilled in order for Medicare coverage to be provided for a VAD used as a bridge to transplant:
- The patient is approved for heart transplantation by a Medicare-approved heart transplant center and is active on the Organ Procurement and Transplantation Network (OPTN) heart transplant waitlist.
- The implanting site, if different than the Medicare-approved transplant center, must receive written permission from the Medicare-approved transplant center under which the patient is listed prior to implantation of the VAD.
3. Destination Therapy (effective for services performed on or after October 1, 2003)
Destination therapy is for patients that require permanent mechanical cardiac support. The VADs used for destination therapy are covered only if they have received approval from the FDA for that purpose.
Patient Selection (effective November 9, 2010):
The VADs are covered for patients who have chronic end-stage heart failure (New York Heart Association Class IV end-stage left ventricular failure) who are not candidates for heart transplantation, and meet the following conditions:
- Have failed to respond to optimal medical management (including beta-blockers and ACE inhibitors if tolerated) for 45 of the last 60 days, or have been balloon pump-dependent for 7 days, or IV inotrope-dependent for 14 days; and,
- Have a left ventricular ejection fraction (LVEF) <25%; and,
- Have demonstrated functional limitation with a peak oxygen consumption of < 14 ml/kg/min unless balloon pump- or inotrope-dependent or physically unable to perform the test.
Facility Criteria (effective XXX):
Facilities implanting a VAD as destination therapy must, at a minimum, meet the following criteria:
Patients must be cared for by a team defined as a cohesive, multidisciplinary team of medical professionals with the appropriate qualifications, training and experience. The team embodies collaboration and dedication across medical specialties to offer optimal patient-centered care. Collectively, the team must ensure that patients and caregivers have the knowledge and support necessary to participate in shared decision making and to provide appropriate informed consent. The team members must be based at the facility and must include individuals with experience working with patients before and after placement of a VAD.
The team must include, at a minimum:
- At least one physician with cardiothoracic surgery privileges and individual experience implanting at least 10 VADs over the course of the previous 36 months with activity in the last year.
- At least one cardiologist trained in advanced heart failure with clinical competence in medical- and device-based management including VADs, and clinical competence in the management of patients before and after heart transplant.
- A VAD program coordinator.
- A social worker.
- A palliative care specialist.
Facilities must track patient outcomes including survival, adverse events (e.g. bleeding, infection, stroke, and device malfunction), functional status, and quality of life in a way that allows comparisons with other institutions and facilitates internal quality monitoring and improvement.
Facilities must be credentialed by an organization approved by CMS.
C. Nationally Non-Covered Indications
All other indications for the use of VADs not otherwise listed remain non-covered, except in the context of Category B investigational device exemption clinical trials (42 CFR 405) or as a routine cost in clinical trials defined under section 310.1 of the National Coverage Determinations (NCD) Manual.
D. Other
This policy does not address coverage of VADs for right ventricular support, biventricular support, use in patients under the age of 18, or use in patients with complex congenital heart disease. Coverage for items and services under section 1862(a)(1)(A) of the Social Security Act in these situations will be made by local Medicare Administrative Contractors (MACs) within their respective jurisdictions.
(This NCD last reviewed XXX.)


