To: Administrative File: CAG-00431N
Beta Amyloid Positron Emission Tomography in Dementia and Neurodegenerative Disease
From: Louis Jacques, MD
Director, Coverage and Analysis Group
Tamara Syrek Jensen, JD
Deputy Director, Coverage and Analysis Group
James Rollins, MD, PhD
Division Director
Brijet Burton Coachman, MPP, MS, PA-C
Lead Analyst
Stuart Caplan, RN, MAS
Analyst
Rosemarie Hakim, PhD
Epidemiologist
Jeffrey Roche, MD, MPH
Medical Officer
Joseph Hutter, MD, MA
Lead Medical Officer
Subject: Final Decision Memorandum for: CAG-00431N
Beta Amyloid Positron Emission Tomography in Dementia and Neurodegenerative Disease
Date: September 27, 2013
I. Final Decision
A. The Centers for Medicare & Medicaid Services (CMS) has determined that the evidence is insufficient to conclude that the use of positron emission tomography (PET) amyloid-beta (Aβ) imaging is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member for Medicare beneficiaries with dementia or neurodegenerative disease, and thus PET Aβ imaging is not covered under §1862(a)(1)(A) of the Social Security Act (“the Act”).
B. However, there is sufficient evidence that the use of PET Aβ imaging is promising in two scenarios: (1) to exclude Alzheimer’s disease (AD) in narrowly defined and clinically difficult differential diagnoses, such as AD versus frontotemporal dementia (FTD); and (2) to enrich clinical trials seeking better treatments or prevention strategies for AD, by allowing for selection of patients on the basis of biological as well as clinical and epidemiological factors.
Therefore, we will cover one PET Aβ scan per patient through coverage with evidence development (CED), under §1862(a)(1)(E) of the Act, in clinical studies that meet the criteria in each of the paragraphs below.
Clinical study objectives must be to (1) develop better treatments or prevention strategies for AD, or, as a strategy to identify subpopulations at risk for developing AD, or (2) resolve clinically difficult differential diagnoses (e.g., frontotemporal dementia (FTD) versus AD) where the use of PET Aβ imaging appears to improve health outcomes. These may include short term outcomes related to changes in management as well as longer term dementia outcomes.
Clinical studies must be approved by CMS, involve subjects from appropriate populations, and be comparative and longitudinal. Where appropriate, studies should be prospective, randomized, and use postmortem diagnosis as the endpoint. Radiopharmaceuticals used in the PET Aβ scans must be FDA approved. Approved studies must address one or more aspects of the following questions. For Medicare beneficiaries with cognitive impairment suspicious for AD, or who may be at risk for developing AD:
- Do the results of PET Aβ imaging lead to improved health outcomes? Meaningful health outcomes of interest include: avoidance of futile treatment or tests; improving, or slowing the decline of, quality of life; and survival.
- Are there specific subpopulations, patient characteristics or differential diagnoses that are predictive of improved health outcomes in patients whose management is guided by the PET Aβ imaging?
- Does using PET Aβ imaging in guiding patient management, to enrich clinical trials seeking better treatments or prevention strategies for AD, by selecting patients on the basis of biological as well as clinical and epidemiological factors, lead to improved health outcomes?
Any clinical study undertaken pursuant to this national coverage determination (NCD) must adhere to the timeframe designated in the approved clinical study protocol. Any approved clinical study must also adhere to the following standards of scientific integrity and relevance to the Medicare population.
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants’ health outcomes.
- The research study is well supported by available scientific and medical information or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it must be in compliance with 21 CFR parts 50 and 56.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed here as Medicare requirements.
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject.
- The research study protocol specifies the method and timing of public release of all pre-specified outcomes to be measured including release of outcomes if outcomes are negative or the study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations on the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with §1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
All other uses are noncovered.
II. Background
Definitions
The following radiopharmaceuticals are referenced in this decision memorandum (DM):
- Florbetapir is florbetapir F18 (or AV-45)
- Florbetaben is florbetaben F18 (or AV-1, or BAY-94-9172)
- Flutemetamol is flutemetamol F18 (or GE-067)
- FDDNP is FDDNP F18
- AZD4694 is AZD4694 F18 (or NAV4694)
- PIB is Pittsburgh Compound B C11
- FDG is fluoro-D-glucose F18
The terms “PET Aβ imaging,” “amyloid-beta PET,” “PET Aβ,” “amyloid imaging,” “amyloid PET,” “Aβ imaging,” “amyloid-beta imaging” and “beta-amyloid imaging” are used synonymously in the literature and in this DM.
Dementia
Dementia is a syndrome involving cognitive and behavioral impairment in an otherwise alert patient, due to a number of neurological diseases, alone or combined. It is not a specific cause or disease process itself. The impairment must involve a minimum of two domains (memory, reasoning, visuospatial abilities, language or personality behaviors); impact daily functioning; represent a decline from previous levels of functioning; not be explainable by delirium (a temporary state of mental confusion and fluctuating consciousness from various causes) or a major psychiatric disorder; and be objectively documented by a “bedside” mental status exam (e.g., the mini-mental status exam) or neuropsychological testing (McKhann 2011).
Mild cognitive impairment (MCI)
Increasingly, research has focused on early stages of cognitive impairment, which lie between the cognitive changes of normal aging and dementia. Mild cognitive impairment (MCI) is a syndrome in which persons experience memory loss (amnestic MCI) or loss of thinking skills other than memory loss (non-amnestic MCI), to a greater extent than expected for age, but without impairment of day-to-day functioning. The clinical work up for MCI is similar to that for AD and other causes of dementia (discussed below).
Individuals with MCI are at increased risk of developing dementia (whether from AD or another etiology), but many do not progress to dementia, and some get better. MCI has multiple subtypes, discussed in more detail later in this DM. These subtypes, and associated results from “bedside” mental status exams and neuropsychiatric testing, could, when combined with (1) other patient characteristics (e.g., age, genetics, cognitive reserve, comorbidities), and (2) biomarkers (for hypometabolism, plaque accumulation, synaptic dysfunction and neuronal loss), serve as the foundation for the development of objectively defined “risk pools,” or subpopulations of individuals who are at risk of progressing from MCI or even pre-symptomatic states to AD (Petersen 1999 and 2009, Wolk 2009, Hughes 2011, Ward 2012, Landau 2012, Sachdev 2012).
Alzheimer’s disease (AD)
Epidemiology, clinical criteria, causes and treatment
AD is an irreversible dementia characterized by progressive, relentless cognitive and functional decline. It is the number one cause of dementia in older Americans (age 65 and over), contributing to 60-80% of cases. Over 5 million older Americans (> 12.5%) have AD. This prevalence is expected to rise to 8.7 million by 2030, and could reach 13.8 million by 2050. AD is the 5th leading cause of death in older Americans (and the 7th leading cause of death overall). Older African-Americans are two times as likely to have AD (and other dementias) as older whites. Older Hispanics are 1.5 times as likely to have AD as older whites. Women are more likely to have AD than men, although this is in part because women live longer (NIA 2013, Brookmeyer 2011, CDC 2013, AA 2013).
Clinical criteria for diagnosing AD are informed by the NIA-AA 2011guidelines (McKhann 2011). Core clinical criteria for “probable AD” dementia must first meet the criteria for “all-cause” dementia described above. Additionally, there must be: (a) insidious onset; (b) documented worsening of cognition; (c) exclusion of major concomitant cerebrovascular disease (as most individuals with AD have some level of this as well); and (d) exclusion of alternative diagnoses (such as dementia with Lewy bodies (DLB), behavioral variant frontotemporal dementia (FTD), progressive aphasia or other neurological disease associated with dementia). A clinical diagnosis of “possible AD” dementia would meet the criteria for “probable AD” above, with the exception of having an “atypical course” (e.g., sudden rather than insidious onset) or an “etiologically mixed presentation.”
The first symptom of AD is usually memory loss (amnesia), due to synaptic dysfunction and loss of neurons in the hippocampus. This leads to impairment of reasoning, judgment, behavior and communication, as well as motor functions, as the disease spreads to other regions of brain. Rarely the initial (or “presenting”) symptoms can be nonamnestic, such as disturbances in language, visuospatial abilities or decision-making.
Most individuals with AD become symptomatic after age 60. Generally an indolent process, it is typically fatal within 8-10 years of onset but can be fatal anywhere between 2 and 20 years. Among 70-year-olds, 61% of those with AD die within a decade (compared to only 30% of those without AD) (NIA 2013, Dilworth 2008, AA 2013).
The underlying cause of AD remains unknown. The number one risk factor is age itself. Investigators hypothesize that a wide range of factors may contribute to its development, including genetic, metabolic, inflammatory, mitochondrial, environmental, and neuronal, to include both cytoskeletal (within the neuronal cell itself) and synaptic (the connectivity among cells) (ECRI 2012, Pimplikar 2010, Herrup 2010, Sperling 2011).
Currently, there is no effective treatment for AD. Existing interventions do not prevent, modify or cure the disease process. Some medications, such as memantine and cholinesterase inhibitors, can temporarily improve cognitive and neuropsychiatric symptoms in some patients with AD (as well as certain other dementias). Care is therefore primarily supportive and increases as functional impairment progresses, eventually leading to round-the-clock supervision which can be needed for years.
Diagnostic work-up, integration of biomarkers, and their shortcomings
The clinical work-up for patients presenting with symptoms of dementia or cognitive impairment, including MCI with possible AD, is extensive. It includes a medical history taken from the patient and from an informant who is well acquainted with the affected person, a physical examination comprising a mental status evaluation aided by quantitative scales and/or neuropsychological assessment, and laboratory testing and often structural neuroimaging such as MRI or CT to rule out other diseases. Clinical assessment is performed primarily using two sources: the National Institute on Aging and the Alzheimer’s Association (NIA-AA) 2011 criteria, which updates the NINCDS-ADRA 1984 criteria to “incorporate more modern innovations in clinical imaging and laboratory assessment” (McKhann 2011); and the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) criteria for dementia of the Alzheimer’s type.
The innovations in “imaging and laboratory assessment” above refer to biomarkers. There are two types: those detecting amyloid-beta (Aβ) protein deposition; and those detecting downstream neuronal degeneration or injury (Jack 2011). Examples of the former type include: direct imaging of amyloid plaques in living brain with florbetapir, PIB and other agents; and decreased Aβ42 in cerebral spinal fluid (CSF), resulting from accumulation of this molecule in the brain. Examples of the latter type include: atrophy of hippocampus and entorhinal cortex on MRI, reflecting neuronal loss; increased total tau protein in CSF, which correlates with neuronal damage; and increased phosphorylated-tau (p-tau) in CSF, which correlates with formation of neurofibrillary tangles (NFTs) (Jack 2008, Sperling 2011, Hampel 2008, Mattsson 2009).
This distinction between amyloid deposition and neuronal degeneration becomes important in current theories of the role of amyloid in the development of AD (discussed below). Increasing use of biomarkers in clinical research has given rise to two new proposed classifications for AD in the NIA-AA 2011 criteria: “probable” or “possible” AD dementia “with evidence of AD pathophysiology.”
These proposed classifications are explicit hypotheses to be assessed through further research. Currently, there are no established biological or neuroimaging markers for the diagnosis of AD or related disorders. Accordingly, the NIA-AA workgroup on dementia concludes that “the core clinical criteria for AD dementia will continue to be the cornerstone of the diagnosis in clinical practice, but biomarker evidence is expected to enhance the pathophysiological specificity of the diagnosis of AD dementia. Much work lies ahead for validating the biomarker diagnosis of AD dementia” (McKhann 2011).
Unfortunately, despite being the “cornerstone” of diagnosis, clinical assessment of AD remains poor. For example, a review of 919 subjects with both clinical and neuropathologic (autopsy) data collected from the NIA-sponsored National Alzheimer’s Coordinating Center Uniform Data Set between 2005-2010 demonstrated sensitivity of clinical diagnosis ranging from 70.9% to 87.3%, and specificity ranging from 44.3% to 70.8% (depending on the restrictiveness of the clinical criteria); this study also found that 39% of subjects with dementia not clinically diagnosed with AD actually had “minimum levels of AD histopathology” (Beach 2012). Other studies found the clinical diagnosis of AD by expert neurologists to be 81% sensitive and 70% specific compared to neuropathology (Knopman 2001, Grundman 2012).
Clinical diagnosis is poor because several other neurological diseases can mimic the dementia seen in AD, including cerebrovascular dementia, dementia with Lewy bodies (DLB), behavioral variant frontotemporal dementia (FTD), Parkinson’s disease, Creutzfeld-Jakob disease, and normal pressure hydrocephalus (NPH). Accordingly, NIA-AA 2011 guidelines require exclusion of these diseases as one of the criteria for clinical diagnosis of “probable AD.” Also, one or more of these diseases, most commonly vascular disease, co-exist in the majority of individuals with AD, as seen at autopsy (Schneider 2007). So there are relatively few patients with “pure” AD. Finally, it is not possible to measure the partial contributions of various coexisting diseases, identified either during life with imaging or biomarkers, or at autopsy, to a patient’s symptoms of dementia.
Pathophysiology and the diagnostic gold standard for AD
The pathophysiological hallmarks of AD are Aβ plaques, neurofibrillary tangles (NFTs) of the protein tau, and neuronal dysfunction and loss. However, amyloid plaques are seen in other diseases, such as dementia with Lewy bodies, cerebral amyloid angiopathy, Parkinson’s disease, Huntington’s disease, and inclusion body myositis. Amyloid plaques can also be detected in cognitively normal older adults. Autopsy studies demonstrate that approximately 33% of older individuals (20-65% depending on age) who are cognitively normal have amyloid accumulation at levels consistent with AD pathology (Hulette 1998, Price 1999, Knopman 2003, Rowe 2010). Finally, amyloid is associated with physiologic processes of disease prevention or response, such as protection against oxidative stress, regulation of cholesterol transport, and anti-microbial activity (Guglielmotto 2010, Zou 2002, Yao 2002, Soscia 2010).
Because clinical diagnosis is poor, and amyloid pathology is seen in other diseases as well as in cognitively normal older persons, the “gold standard” for diagnosis requires both (a) the presence of moderate to frequent Aβ plaques and neurofibrillary tangles on autopsy, and (b) clinical documentation of progressive dementia during life (NIA-Reagan Institute 1997, Hyman 1997).
Competing views on the role of amyloid
Acknowledging that there are competing views on the role of amyloid in the pathophysiology of AD is key to interpreting the significance of trials on AD prognosis, diagnosis and clinical utility. It is widely accepted that the presence of amyloid plaques in human brain is virtually necessary for the diagnosis of AD. It is built into the postmortem diagnostic gold standard, and reflected in the FDA-approved label for florbetapir (Sperling 2011, NIA-Reagan 1997, FDA 2012). However, whether a threshold level of amyloid plaques in a patient is sufficient for diagnosing AD is a subject of much debate. One hypothesis is that patients with symptoms of cognitive impairment and evidence of brain amyloid have AD, and it is just a matter of time before this manifests clinically as AD dementia.
A competing hypothesis is that “Aβ accumulation is necessary but not sufficient to produce the clinical manifestations of AD. It is likely that the cognitive decline would occur only in the setting of Aβ accumulation plus synaptic dysfunction and/or neurodegeneration” (Sperling 2011).
In this light, the NIA-AA criteria authors conclude that “at this point, it remains unclear whether it is meaningful or feasible to make the distinction between Aβ as a risk factor for developing the clinical syndrome of AD versus Aβ accumulation as an early detectable stage of AD because current evidence suggests that both concepts are plausible” (Sperling 2011).
PET Aβ imaging
PET is a minimally invasive diagnostic imaging procedure used to evaluate normal tissue as well as diseased tissues in conditions such as cancer, ischemic heart disease and some neurologic disorders. A ligand that binds to a given targeted substrate (e.g., Aβ plaque aggregates) is labeled with a radioisotope (e.g., fluorine F18). The injected radiopharmaceutical (or “tracer”) emits positrons when it decays. PET uses a positron camera (tomograph) to measure the decay of such tracers within human tissue. The relative differences in the rate of tracer decay among anatomic sites provide biochemical information on the tissue being studied.
PET Aβ imaging detects amyloid plaque density in vivo in human brain. While several Aβ imaging agents exist, including Pittsburg compound B (PIB C11), and several F18 labeled agents (florbetapir; florbetaben; flutemetamol; AZD469; and FDDNP, which images both amyloid and tau), the longer half-lives of the F18-labelled agents render them more practical in clinical settings. As the only FDA-approved agent for PET Aβ imaging to date is florbetapir, it is the primary focus of our review.
III. History of Medicare Coverage
CMS did not previously cover PET Aβ imaging. FDG PET is nationally covered for either the differential diagnosis of FTD versus AD under specific requirements; or, its use in a CMS-approved practical clinical trial focused on the utility of FDG PET in the diagnosis or treatment of dementing neurodegenerative diseases. FDG PET for dementia and neurodegenerative diseases and other specific covered uses of particular PET radioactive tracers (N13 ammonia, Rb82 and F18 sodium fluoride (NaF-18) are found in detail in Section 220.6 of the National Coverage Determination Manual available at http://www.cms.gov/Regulations-and-Guidance/Guidance/Manuals/Downloads/ncd103c1_Part4.pdf.
A. Current Request
In July 2012 Lilly USA, LLC, manufacturer of the PET amyloid radiopharmaceutical florbetapir (Amyvid™), requested that CMS reconsider its non-coverage decision for PET scans and provide coverage for the use of PET amyloid imaging as a diagnostic test to “estimate amyloid neuritic plaque density in adult patients with documented cognitive impairment who are being evaluated for Alzheimer’s disease (AD) and other causes of cognitive impairment” (Requestor Letter, at http://www.cms.gov/medicare-coverage-database/details/nca-tracking-sheet.aspx?NCAId=265&fromdb=true).
B. Benefit Category
Medicare is a defined benefit program. An item or service must fall within a benefit category as a prerequisite to Medicare coverage (§1812 (Scope of Part A); §1832 (Scope of Part B) and §1861(s) (Definition of Medical and Other Health Services) of the Act. PET is considered to be within the following benefit category: other diagnostic tests §1861(s)(3) of the Act).
IV. Timeline of Recent Activities
| Date |
Action |
| October 9, 2012 |
CMS accepts the formal request for the coverage of PET Aβ imaging in the diagnosis of AD and other causes of cognitive decline. A 30-day public comment period begins. |
| November 8, 2012 |
The 30-day public comment period ends. CMS received 27 timely comments. |
| July 3, 2013 |
CMS posts the proposed decision memorandum for 30 days of public comment. |
| August 2, 2013 |
The public comment period on the proposed decision memorandum closes with 202 comments received. |
V. FDA Status
The FDA has reviewed and approved one radiopharmaceutical for PET Aβ imaging, florbetapir (Amyvid™), in April 2012, to estimate Aβ neuritic plaque density in adult patients with cognitive impairment who are being evaluated for AD and other causes of cognitive decline. In the FDA-approved label for florbetapir there is no definition of “cognitive impairment,” but the label does reference studies whose cognitively impaired patient populations range from MCI to dementia. The label states that although a negative florbetapir scan reduces the likelihood of AD, a positive florbetapir scan does not confirm the diagnosis of AD or any other cognitive disorder. This is because a positive florbetapir scan, which indicates the presence of moderate to frequent amyloid plagues in the brain, may be seen in persons with AD or other causes of cognitive decline as well as in persons with normal cognition.
The FDA-approved label for florbetapir indicates that it was not evaluated by the FDA as a screening tool to predict the development of dementia (including AD) or other cognitive disorders, nor to monitor the therapeutic response to treatment of these neurological conditions. Additionally, the label indicates that florbetapir images should only be interpreted by readers who successfully complete a special training program, which has been provided by the manufacturer through an in-person tutorial or electronic process. The FDA-approved label for florbetapir can be viewed in its entirety at http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202008s000lbl.pdf
VI. General Methodological Principles
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether the evidence is sufficient to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: (1) the specific assessment questions can be answered conclusively; and (2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary. A detailed account of the methodological principles of study design that CMS uses to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public commenters sometimes cite the published clinical evidence and provide CMS with useful information. Public comments that provide information based on unpublished evidence, such as the results of individual practitioners or patients, are less rigorous and, therefore, less useful for making a coverage determination. CMS uses the initial comment period to inform its proposed decision. CMS responds in detail to the public comments that were received in response to the proposed decision when it issues the final decision memorandum.
VII. Evidence
A. Introduction
The purpose of this evidence review is to summarize the published literature on whether PET Aβ imaging is beneficial to patients with symptoms of AD. The evidence reviewed here includes the published medical literature as of August 31, 2013, on pertinent clinical trials, focusing on florbetapir, as it is the only clinically-relevant, FDA-approved PET Aβ imaging tracer. Additional supporting evidence from other studies and sources are cited below.
B. Summary of Evidence
1. Questions:
- Is the evidence adequate to conclude that PET Aβ imaging improves meaningful health outcomes in beneficiaries who display signs or symptoms of AD?
- Is the evidence adequate to conclude that PET Aβ imaging results inform the treating physician's management of the beneficiary to improve meaningful health outcomes? Those outcomes may include reasonably considered beneficial therapeutic management or the avoidance of unnecessary, burdensome interventions.
2. External Technology Assessment
CMS did not request an external technology assessment (TA) on this issue.
3. Internal technology assessment
Literature search methods
Literature searches performed on PubMed included combinations of the following terms: amyloid, beta-amyloid, PET imaging, dementia, Alzheimer’s disease, neurodegenerative disorders, and mild cognitive impairment. Searches were also performed, using the same search terms, in ClinicalTrials.gov, the National Guideline Clearinghouse, the Cochrane Library, EMBASE, and other sources such as Trip Database.
Additional articles were selected from citations from key clinical trials, recent review articles, the NCD request, expert speaker talks at the MEDCAC meeting, MEDCAC panel members and public comments.
A review of the medical literature failed to reveal any pertinent meta-analysis or systematic reviews evaluating specifically the use of PET Aβ imaging in patients with signs and symptoms of AD. Although no randomized clinical trials were found exploring the use of PET Aβ imaging in this population, most studies found were prospective longitudinal studies. One study employed the use of a cross-sectional design (Landau 2012).
Prospective Longitudinal Studies
Wong D, Rosenberg P, Zhou Y, Kumar A, Raymont V, Ravert H, et al. In Vivo Imaging of Amyloid Deposition in Alzheimer’s Disease using the Novel Radioligand [18F]AV-45 (Florbetapir F 18). J Nucl Med. 2010 June;51(6):913–920.
Wong and associates performed a study designed to explore brain imaging properties in cognitively healthy patients and those with AD by using PET florbetapir imaging. This open-label, multicenter, study involved 16 patients with Alzheimer’s disease, as well as 16 cognitively healthy controls; both groups received florbetapir and PET imaging (in AD patients the mean age was 75.8 +/- 9.2, in healthy controls (HC) the mean age was 72.5 +/- 11.6). Patients with AD had to be greater than 50 years of age and have a probable diagnosis of AD according to NINCDS-ADRDA criteria, with a mini-mental status examination (MMSE) score between 10 and 24 inclusive. All healthy control subjects also had to be greater than 50 years of age, have no evidence of cognitive impairment by history and psychometric testing, and had to have an MMSE score of ≥ 29. Subjects who showed evidence of any other significant neurodegenerative or psychiatric disease on clinical examination or MRI, or clinically significant medical comorbidities, were excluded from the study. In the study, standard uptake values ratios (SUVR) were calculated using cerebellar grey matter as the primary reference region, and centrum semiovale white matter as an alternative reference region, and a parametric mapping approach employing the cerebellum as a reference region was used to calculate distribution/volume ratios (DVR).
Looking at the demographics of the two groups, though the baseline average MMSE was lower in the AD subjects than in the HC subjects (19.1 +/− 3.1 vs. 29.8 +/− 0.45), both groups were similar in age, weight, and education. A review of baseline data also revealed that there were a slightly higher proportion of males in the healthy control group than in the AD group (10/16 versus 8/16, respectively).
Results of the study revealed that accumulation of florbetapir tracer was found in cortical target areas such as the frontal cortex, temporal cortex and precuneus, areas that were expected to be high in amyloid deposition, while in healthy control subject tracer accumulation predominantly was distributed in the white matter areas. The cortical to cerebellar SUVR values remained much longer in AD patients than in healthy controls, reaching a plateau within 50 minutes. Using the 10 minute period from 50–60 minutes post administration as a representative sample, the cortical average SUVR for this period was 1.67 +/− 0.175 for patients with AD vs. 1.25 +/− 0.177 for healthy control subjects. The study also revealed that spatially normalized DVRs generated from PET dynamic scans were highly correlated with SUVR (r = 0.58–0.88, p < 0.005) and were significantly greater for AD patients than for healthy control subjects in cortical regions, but not in subcortical white matter or cerebellar regions.
The authors concluded that florbetapir PET imaging showed significant discrimination between clinically diagnosed AD patients and healthy control subjects using either a parametric reference region method (DVR) or a simplified SUVR method.
Camus V, Payoux P, Barré L, Desgranges B, Voisin T, Tauber C, et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur J Nucl Med Mol Imaging. 2012 Apr;39(4):621-31. doi: 10.1007/s00259-011-2021-8. Epub 2012 Jan 18.
Camus and associates performed a prospective study to evaluate the clinical usefulness of florbetapir. The purpose of the study was to assess the feasibility of using PET imaging with florbetapir in three-level clinical settings to differentiate patients with mild to moderate AD or MCI patients from normal healthy control subjects in three PET centers. They also wanted to assess the safety of a florbetapir injection immediately after injection and during the follow-up period. Subjects included consecutive patients referred from the three participating memory clinics associated with the study center in France, and who met specific criteria as stated in the NINCDS-ADRDA criteria set for probable AD and DSM-IV criteria for Alzheimer’s type dementia or diagnostic criteria for amnestic MCI. All participants had to be at least 55 years of age, be able to speak French fluently, have completed at least seven years of education and have neither unstable somatic disease nor psychiatric comorbidities. Healthy subjects who acted as controls were recruited through a community advertisement and evaluated in the same clinical settings.
The diagnosis of AD was confirmed using a mini-mental state examination (MMSE), as well as meeting the guidelines for global neuropsychological testing and an evaluation of verbal episodic memory (Free and Cued Selective Reminding Test, FCSRT), language (verbal fluency, naming, comprehension), gnosis, praxis, visuospatial functions and executive functions. Patients were excluded if they had any past or current symptomatic treatment with acetylcholinesterase inhibitors or memantine or had participated in any experimental study investigating Aβ-lowering agents. For MCI patients, a subjective memory complaint associated with isolated impairment in episodic memory had to be present, and assessed by a free recall total based on FCSRT. Healthy controls used in the study could not have any past history of or current major depressive episodes and/or antidepressant treatment, cognitive impairment in the diagnostic neuropsychological battery, memory complaints, or MRI brain scan abnormalities. A total of 46 subjects (20 men, 26 women, mean age 69.0 ± 7.6 years) were included in the study, including 13 AD patients, 12 MCI patients and 21 healthy control subjects. A brain MRI scan, a whole-body hybrid PET/CT scan and florbetapir PET imaging was performed on all subjects. PET images were assessed visually by blinded inspectors to any clinical information and quantitatively via the standard uptake value ratio (SUVR) in the specific regions of interest, which were defined in relation to the cerebellum as the reference region.
Results of the study revealed that the PET scan procedures were well tolerated, and no serious adverse events were reported during the immediate follow-up period, though at the 1-year follow-up, two patients did had medical problems unrelated to the study and were excluded from the analysis. The mean values of SUVR were higher in AD patients (median 1.20, Q1-Q3 1.16-1.30) than in healthy control subjects (median 1.05, Q1-Q3 1.04-1.08; p = 0.0001) in the overall cortex and in all cortical regions (precuneus, anterior and posterior cingulate, and frontal median, temporal, parietal and occipital cortex). The MCI subjects also showed a higher uptake of florbetapir in the posterior cingulate cortex (median 1.06, Q1-Q3 0.97-1.28) compared with healthy control subjects (median 0.95, Q1-Q3 0.82-1.02; p = 0.03). Qualitative visual assessment of the PET scans showed a sensitivity of 84.6% (95% CI 0.55 – 0.98) and a specificity of 38.1% (95% CI 0.18 – 0.62) for discriminating clinically diagnosed AD patients from healthy control subjects; however, the quantitative assessment of the global cortex SUVR showed a sensitivity of 92.3% and specificity of 90.5% with a cut-off value of 1.122 (area under the curve 0.894).
Based on the results of the study, the authors felt that PET with florbetapir was suitable for routine use to improve the accuracy of AD diagnosis in the clinical setting, because the quantitative analyses showed a higher global SUVR and SUVR in several cortical regions (precuneus, anterior and posterior cingulate, frontal median, temporal, parietal and occipital cortex) in AD patients than in healthy control subjects. It also showed that the SUVR in the posterior cingulate and frontal median regions was significantly higher in AD patients than in MCI patients. The authors also note the following:
- the pattern of florbetapir cortical uptake found in the present study is similar to that found in previous studies conducted by Wong et al. and Clark et al.;
- the pattern also appears to be similar to those found with other amyloid-labeling compounds, such as PIB C11 and its flutemetamol F18-derived molecule, 11C-BF-227, FDDNP F18 and BAY94-9172 F18; and
- these patterns closely match the neuropathological stages of AD progression, which was strengthened by the high correlation found between florbetapir PET imaging and autopsy results.
The authors concluded that PET with florbetapir should become a routine clinical procedure because it improves the reliability of AD diagnosis and the detection of typical or atypical forms of pre-dementia stages, such as amnestic MCI and MCI associated with multi-domain deficits or neuropsychiatric symptoms (e.g., depression). But the authors also note that more studies testing the feasibility and tolerability of consecutive scans with florbetapir are needed to better document the accuracy of PET imaging with florbetapir in the AD diagnostic process at the dementia or pre-dementia stages, and that comparisons (or combinations) with other biomarkers, such as FDG PET, MRI and CSF dosages of tau and protein, are also needed.
Clark CM, Sneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA. Use of Florbetapir PET for Imaging Aβ Pathology. JAMA 2011 Jan 19;305(3):275-83.
Clark and associates performed a prospective clinical evaluation study to determine the qualitative and quantitative relationship between the florbetapir PET image and postmortem-amyloid pathology. This phase 3 multicenter study had two cohort groups. One group involved individuals at the end of life who consented to both florbetapir PET imaging and brain donation after death. In the other group, PET images were also obtained from younger individuals presumed to be free of brain amyloid to better understand the frequency of a false positive florbetapir PET image.
The study enrolled 152 individuals who were at least 51 years of age and approaching the end of their life, to obtain 35 postmortem brain evaluations from those who received PET imaging 12 months or less prior to death. Inclusion criteria for this group included a physician’s assessment that the individual was likely to die within six months of study enrollment, absence of any known destructive lesion in the brain (e.g., stroke or tumor), and the individual’s willingness to have florbetapir PET imaging followed by a brain autopsy at the time of death. The study also involved a second group of 74 young, cognitively normal, healthy individuals (aged 18-50 years). In both groups, physical, neurological, and cognitive evaluations that included assessments of memory, language, and constructional praxis were obtained.
Participants were imaged at 23 sites using clinical PET and PET/computed tomographic scanners, and florbetapir PET images were visually assessed by three board-certified nuclear medicine physicians, using a semi-quantitative score ranging from 0 (no amyloid) to 4 (high levels of cortical amyloid). A semi-automated quantitative analysis of the ratio of cortical to cerebellar signal (SUVR) also was performed for florbetapir PET images from all study participants. The main outcome measure of the study was correlation of florbetapir PET image interpretation (based on the median of 3 nuclear medicine physicians’ ratings) and semi-automated quantification of cortical retention with postmortem Aβ burden, neuritic amyloid plaque density, and neuropathological diagnosis of Alzheimer disease in the first 35 participants autopsied (out of 152 individuals enrolled in the PET pathological correlation study). Autopsied brain tissue was obtained to identify and quantify Aβ aggregation using an automated immunostainer following established immunohistochemistry methods, and PET image quantification was performed using image processing and analysis software. Aβ neuritic plaque density was determined, and the mean density for both neuritic and diffuse plaques, using silver stain, was summarized by anatomical region using a 4-point semi-quantitative scale (0 = none, 1 = sparse, 2 = moderate, 3 = severe). Also, a neuropathological diagnosis was made using standardized criteria as described by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) and the National Institute on Aging (NIA) and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (NIA/Reagan Institute criteria).
Results of the study revealed that there were significant correlations between the two measures of amyloid on florbetapir PET (SUVR versus semiquantitative visual score: 0.82 [95% CI, 0.64 - 0.87]; p < .001) and the two measures of amyloid aggregation at autopsy (immunohistochemistry vs. silver stain: 0.88 [95% CI, 0.76 - 0.94]; p < .001). The strengths of the inter-method correlations (e.g., PET visual read to immunohistochemistry) were similar to that for the intra-method correlations (e.g., PET visual read to PET SUVR, pathology immunohistochemistry to pathology plaque score). The study also revealed that 15 participants in the primary analysis autopsy cohort met pathological criteria for AD (CERAD: probable or definite AD; NIA/Reagan Institute criteria: intermediate to high likelihood of AD) and of these 15 participants, 14 had florbetapir PET scans that were interpreted as visually positive (median read 2), giving a sensitivity of 93% (95% CI, 68% - 100%). Finally, 14 participants in the autopsy cohort had low levels of Aβ aggregation on the postmortem examination and did not meet CERAD or NIA/Reagan Institute pathological criteria for AD. All 14 had florbetapir PET scans that read as negative, yielding a specificity of 100% (95% CI, 76.8% - 100%). The authors noted that the reviewers who read results for the florbetapir PET images agreed with the final autopsy with respect to the presence or absence of neuropathological criteria of AD in 28 of 29 cases.
The authors concluded that florbetapir PET imaging performed during life in this study correlated with the presence and density of Aβ at autopsy, and felt that this study provides evidence that a molecular imaging procedure can identify Aβ pathology in the brains of individuals during life.
Clark C, Pontecorvo M, Bench T, Bedell B, Coleman R, Doraiswamy P. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic Aβ plaques: a prospective cohort study. Lancet Neural 2012;11:669-78.
This second study by Clark and associates was a continuation of the 2011 discussed above. Like the original study, this prospective cohort study’s purpose was to determine the qualitative and quantitative relationship between florbetapir PET imaging and postmortem-amyloid pathology. Patients who were alive at the end of the first study were followed up to autopsy, or for an additional year after the PET scan. Images and histopathological results from the original cohort study were used and extended to follow-up and were analyzed together to test the diagnostic accuracy of binary visual interpretation of florbetapir PET scans by comparison with the reference standard of neuritic plaque density at autopsy. The original study enrolled 152 individuals and obtained 35 postmortem brain evaluations from those who had received PET imaging 12 months or less prior to death. Autopsy results of the original Clark article was based on this cohort of 35 subjects.
The second Clark study used the same inclusion and exclusion criteria as the original study, as well as the same physical, neurological, and cognitive evaluations that included assessments of memory, language, and constructional praxis. The second study also had three board-certified nuclear medicine physicians read the florbetapir PET images, using a semi-quantitative score ranging from 0 (no amyloid) to 4 (high levels of cortical amyloid). And as before, a semi-automated quantitative analysis of the ratio of cortical to cerebellar signal (SUVR) was performed for florbetapir PET images from all study participants. Autopsied brain tissue was examined to identify and quantify Aβ aggregation, and neuritic plaque density was determined using a 4-point semi-quantitative scale (0 = none, 1 = sparse, 2 = moderate, 3 = severe). The main outcome measure of the study was correlation of florbetapir PET image interpretation and semi-automated quantification of cortical retention with postmortem Aβ burden, and neuritic amyloid plaque density. The neuropathologic diagnosis of AD was made using standardized criteria as described by the CERAD and the National Institute on Aging (NIA) and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease (NIA/Reagan Institute criteria).
In the original Clark study, 35 participants died and had a postmortem exam. The remaining participants were followed up to 1 year, or a maximum of two years after the original PET scan. During this period an additional 24 autopsy results became available, leaving a combined total of 59 participants with a valid florbetapir PET scan and autopsy results within 24 months which comprised the primary efficacy analysis population. The mean age of this group was 79.4 years, and male as well as female genders were equally represented in this study. According to inclusion criteria, 12 subjects had no cognitive impairment, five had mild cognitive impairment that did not meet the criteria for dementia, 29 had AD, and 13 had other forms of dementia (e.g., dementia with Lewy bodies, Parkinson’s disease dementia, frontotemporal dementia, unspecified dementia, and mixed dementia). The secondary efficacy analysis population, which consisted of patients in the 12 month autopsy cohort, had similar demographic and characteristics as the primary efficacy analysis population.
Results of the study revealed that 39 of the 59 patients included in the study in the primary efficacy analysis population had moderate or frequent neuritic plaques at autopsy and were categorized as positive for Aβ according to histopathological assessment. Most readers rated the florbetapir PET scans as positive in 36 of these 39 subjects, giving this a sensitivity rating of 92%. All 20 subjects with no or sparse neuritic plaque at autopsy were categorized as negative by the majority of readers of the florbetapir PET scan, resulting in a specificity of 100%. The overall accuracy for the primary efficacy analysis population was 95%. The sensitivity, specificity, and overall accuracy of the 46 participants included in the secondary efficacy analysis population were 96%, 100% and 98% respectively.
Visual semi-quantitative ratings of Aβ by use of florbetapir PET imaging showed a positive correlation with postmortem levels of Aβ measured via immunohistochemistry in subjects who had autopsies within two years of PET scan (Spearman ρ = 0.76; p < 0.0001), as well as subjects who had autopsies within one year of PET scan (Spearman ρ = 0.79; p < 0.0001). The authors concluded that the results of the study showed correlation between florbetapir PET imaging and postmortem amyloid burden, and the authors concluded that florbetapir might be useful for imaging of Aβ neuritic plaques in the brains of patients with cognitive impairment.
Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N. Using Positron Emission Tomography and Florbetapir F 18 to Image Cortical Amyloid in Patients With Mild Cognitive Impairment or Dementia Due to Alzheimer Disease. Arch Neurol. 2011;68(11):1404-1411.
Fleischer and associates used multiple research imaging centers in their study to characterize quantitative florbetapir PET measurements of fibrillar Aβ burden in a large clinical cohort of participants with probable AD or mild cognitive impairment and older healthy controls. The study used pooled data from the four registered phase I and II trials of florbetapir PET imaging, using standard dosing of florbetapir and non-dynamic PET acquisitions. The study evaluated both continuous and binary measures of florbetapir PET activity to assess global differences between clinical diagnostic groups, to confirm expected patterns of regional distributions of fibrillar Aβ, and to determine proportions of positive scans using cut-off thresholds for global cortical florbetapir activity. During the course of the study, researchers predetermined SUVR threshold levels for defining florbetapir PET positivity based on a previously reported study of expired end-of-life patients and a specificity cohort of young ApoE4 non-carriers.
The study involved a total of 210 participants who were 55 years of age or older, consisting of 82 cognitively normal volunteers, 60 individuals with MCI, and 68 individuals with probable AD. Florbetapir PET scans were taken of all participants, and they were required to have no subjective cognitive complaints as corroborated by an informant report, to have an MMSE score of 29 or greater, and to be cognitively normal based on psychometric testing. Participants with probable AD met NINCDS-ADRDA criteria for probable AD and had an MMSE score at screening in the range of 10 to 24. ApoE genotyping was performed as an optional procedure on 155 participants. Subjects were excluded if they had other current clinically relevant neurologic or psychiatric illnesses, were receiving any investigational medications, or ever received an anti-amyloid experimental therapy.
All participants underwent a florbetapir PET session that consisted of intravenous injection of florbetapir F 18, and a region of interest (ROI) analysis was performed on individual PET images. Cerebral–to–whole-cerebellar florbetapir standard uptake value ratios (SUVRs) were computed. The study compared mean cortical SUVRs, and a threshold of SUVRs greater than or equal to 1.17 was used to reflect pathological levels of amyloid associated with AD based on separate antemortem PET and postmortem neuropathology data from 19 end-of-life patients. Also a threshold of SUVRs greater than 1.08 was used to signify the presence of any identifiable Aβ because this was the upper limit from a separate set of 46 individuals 18 to 40 years of age who did not carry ApoE4. In this study florbetapir PET activity was the outcome measure of interest.
Results of the study revealed that all participant groups differed significantly in terms of mean [SD] cortical florbetapir SUVRs. Those with probable AD had a mean score of 1.39 [0.24], those with MCI had a mean score of 1.17 [0.27], and those who were older healthy controls (HC) had a mean score of 1.05 [0.16] (p < 1.0 x 10−7). In terms of percentage meeting levels of amyloid associated with AD by SUVR criteria the scores were 80.9% (AD), 40.0% (MCI) and 20.7% (HC) (p < 1.0 x 10−7). In terms of percentage meeting SUVR criteria for the presence of any identifiable Aβ the scores were 85.3% (AD), 46.6% (MCI) and 28.1% (HC) (p < 1.0 x 10−7). In older healthy controls, the percentage of florbetapir positivity increased linearly by age decile (p = .05). The study also revealed that for the 54 older health controls with available ApoE genotypes, ApoE4 carriers had a higher mean [SD] cortical SUVR than did non-carriers (1.14 [0.2] versus 1.03 [0.16]; p = .048). The authors felt that the results support the ability of florbetapir PET SUVRs to characterize amyloid levels in
clinically probable AD, MCI, and older healthy control groups, using both continuous and binary quantitative measures of amyloid burden.
Doraiswamy P, Sperling R, Coleman R, Johnson K, Reiman E, Davis, M. Amyloid-β assessed by florbetapir F18 PET and 18-month cognitive decline: A multicenter study. Neurology 2012;79:1636–1644.
Doraiswamy and associates performed a prospective, multicenter, observational study to evaluate the prognostic utility of detecting Aβ pathology using florbetapir PET in older subjects at risk for progressive cognitive decline. In this study, 51 subjects with MCI, 69 clinically normal cognitively healthy controls, and 31 subjects clinically diagnosed with AD dementia who had previously received a florbetapir PET scan were enrolled. Patients with AD dementia met NINCDS-ADRDA criteria for probable AD and had MMSE scores less than or equal to 24. MCI subjects were presenting for an initial evaluation, or had received a diagnosis of MCI within the past year prior to the study. MCI participants had to be at least 50 years of age, had a complaint of memory or cognitive impairment corroborated by an informant, had a clinical dementia rating (CDR) scale global rating of 0.5, and MMSE > 24 and no episodic memory cut-off was required. The healthy control subjects had to be at least 50 years of age, and were assessed clinically as cognitively normal, and had a CDR global of 0 and an MMSE of 29 or 30. Cognitively normal subjects were recruited approximately equally across age deciles (50–59, 60-69, 70–79, and equal to or greater than 80 years of age).
All subjects included in the study underwent a detailed medical history, physical and neurologic examinations, a clinical interview and laboratory evaluations; additionally an MRI was performed at screening or within six months prior to enrollment to rule out significant CNS lesions. Subjects were excluded if they had other relevant neuropsychiatric diseases, received anti-amyloid investigational drugs, were unable to complete psychometric testing, or had contraindications to PET. A battery of procedures was performed on all subjects including a clinical diagnostic interview and cognitive/functional testing including the CDR, MMSE, Alzheimer’s Disease Assessment Scale–Cognitive subscale (ADAS-Cog; 11-item version), Wechsler Logical Memory (immediate and delayed recall), Digit-Symbol Substitution, Category Verbal Fluency (animals and vegetables), Alzheimer’s Disease Cooperative Study–Activities of Daily Living Scale (ADCS-ADL), and Geriatric Depression Scale (GDS). ApoE genotyping was also performed.
Subjects underwent PET amyloid imaging using florbetapir. Three nuclear medicine physicians, blinded to clinical data, independently reviewed all PET images and rated each on both a semi-quantitative (0–4) and a binary qualitative scale (amyloid positive or amyloid negative) based on the pattern of tracer uptake in gray matter cortical areas. Cerebral-to-whole-cerebellar florbetapir standard uptake value ratios (SUVRs) were calculated using whole cerebellum as the reference region. The average of the SUVR across the six cortical target regions was used for analysis. Subjects who completed the initial PET scan were eligible to participate in the follow-up protocol which would determine whether florbetapir PET predicts progressive cognitive impairment at 36 months.
By the end of the study, of the 151 subjects (69 cognitively normal, 51 mild cognitive impairment, 31 AD) who entered the study, 97% of cognitively normal, 90% of MCI, and 87% of AD subjects completed the 18 months follow-up. The analysis revealed that in both MCI and cognitively normal patients, baseline Aβ positive scans were associated with greater clinical worsening on the Alzheimer’s Disease Assessment Scale–Cognitive subscale (ADAS-Cog (p < 0.01) and Clinical Dementia Rating–sum of boxes (CDR-SB) (p < 0.02). Analysis also revealed that MCI Aβ positive scans were associated with greater decline in memory, Digit Symbol Substitution (DSS) and MMSE scores (p < 0.05). And though MCI subjects had higher baseline SUVR, which was correlated with greater subsequent decline on the ADAS-Cog (p < 0.01), CDR-SB (p < 0.03), a memory measure, DSS, and MMSE (p < 0.05), Aβ positive MCI subjects tended to convert to AD dementia at a higher rate than Aβ negative subjects (p < 0.10).
The authors of the study felt that the results demonstrated that florbetapir amyloid imaging confirms that both cognitively normal subjects and subjects with MCI with higher levels of cortical Aβ on PET are at higher risk for future cognitive progression than individuals with lower levels of amyloid, after controlling for age and baseline cognitive performance. They felt that not only did the findings support the use of florbetapir PET as a predictive biomarker of cognitive decline in at-risk subjects, but also that amyloid PET may have predictive value in MCI for developing AD dementia. They concluded that florbetapir PET may help identify individuals at increased risk for progressive cognitive decline.
Grundman M. Pontecorvo M, Salloway S, Doraiswamy P, Fleisher A, Sadowsky C, et al. Potential Impact of Amyloid Imaging on Diagnosis and Intended Management in Patients With Progressive Cognitive Decline. Alzheimer Dis Assoc Disord 2012;00:000–000.
Grundman and associates performed a prospective study to determine the impact of amyloid imaging on the diagnoses and management of patients undergoing evaluation for cognitive decline, more specifically to determine whether knowledge of the presence or absence of moderate to frequent neuritic amyloid plaques, as assessed by a florbetapir PET scan, would alter a physician’s diagnostic thinking and intended patient management. The study consisted of two roughly equal groups of patients: those who had completed a diagnostic evaluation for progressive cognitive decline/impairment within the previous 18 months (group A, n = 110), and those who were currently undergoing an evaluation (group B, n = 119), but presumably were at a point where the physician was interested in obtaining florbetapir PET scan information. For patients in the study undergoing diagnostic evaluation at entry, the investigator had the option of completing the evaluation and enrolling the patient in group A or enrolling the patient in group B and then considering additional evaluations after the PET scan had been obtained. Although there was no requirement that patients had to meet a specific level of cognitive impairment for inclusion in the study, only patients in whom a history of cognitive decline was documented were included. Exclusion criteria included patients who had a previous amyloid imaging scan or previous participation in a clinical trial of an amyloid targeting therapeutic agents (unless they were in the placebo group).
Screening and baseline studies were obtained, which consisted of a medical history including demographic features, history of cognitive decline, and a record of diagnostic tests performed as part of the standard practice clinical evaluation/diagnostic workup. Subjects also underwent the MMSE. The site physicians decided whether or not patients should be placed in group A (completed their diagnostic evaluation) or group B (still undergoing diagnostic evaluation). If the screening visit/pre-scan evaluation indicated a need for additional diagnostic testing, patients were always assigned to group B. At the end of the screening, physicians recorded the current diagnosis (group A), or working diagnosis (group B) for each patient. Diagnoses were classified as either:
- etiology due to AD (or most likely prodromal AD, or MCI due to AD, probable AD, atypical AD, Lewy body disease with AD/amyloid pathology, or mixed dementia with AD);
- non-AD etiology (most likely etiology is not AD, e.g., mild cognitive impairment of uncertain etiology, but not due to AD; or a specific non-AD etiology such as vascular dementia, frontotemporal dementia; Lewy body disease without AD pathology; primary progressive aphasia; metabolic, psychiatric, or medication-induced impairments); or
- indeterminate (syndromic) etiology, (where the clinician could describe a syndrome but could not provide a more specific etiology, e.g., progressive cognitive decline, mild cognitive impairment, or dementia of uncertain etiology).
For all participants in the study, the treating physicians had to provide results of diagnostic testing and a management plan using information available before florbetapir imaging. After subjects received imaging with florbetapir PET, the diagnosis and intended management at baseline were compared with those obtained after receiving the florbetapir PET scan result. For purposes of this study, a change from an indeterminate/uncertain etiology to a specific etiology (such as MCI due to AD) or a change from one etiologic category (due to AD/not due to AD) to the other was considered a change in diagnosis. A change within etiologic category (e.g., MCI due to AD changed to Dementia due to AD) was not considered a change in diagnosis.
A total of 229 subjects (group A, 48%, n = 110; group B, 52%, n = 119) were enrolled in the study and underwent florbetapir PET scans. The mean age of participants was 74.1 ± 8.1 years, 95% of the subjects were white, and 50.2% were male. With the exception of gender (p = 0.0202), there were no significant demographic differences between subjects who had previously completed a workup and diagnosis and those still undergoing a workup. Of the study participants, 36% had dementia, and the remaining 64% had cognitive impairment not at the level of dementia; also 113 subjects were amyloid positive, while 116 were amyloid negative. Analysis of data revealed that after receiving the results of the florbetapir scan, post-scan diagnosis changed in 125 (54.6%) of 229 cases (95% CI, 48.1% - 60.9%). The scan had an impact on the classification for 37% of subjects with a pre-scan diagnosis indicating an etiology due to AD, 66% of subjects with an indeterminate pre-scan diagnosis, and 62% of subjects with a non-AD pre-scan diagnosis.
When looking at changes in confidence in terms of etiologic diagnosis at both the pre-scan and the postscan time points, the mean confidence level significantly increased after florbetapir PET by an average of 21.6% (95% CI, 18.3% - 24.8%; p < 0.0001. And in terms of intended management, there was a change in the overall management plan for 199 (86.9%) of 229 subjects (95% CI, 81.9% - 90.7%), especially when it came to intended medication management as a result of the scan. In 71 (31%) of 229 subjects (95% CI, 25.4% - 37.3%) florbetapir PET results led to an intended change in AD medications and in 17 (7.4%) of 229 patients (95% CI, 4.7% - 11.6%), the results led to an intended change in treatment with psychiatric medications (e.g., antidepressants, antianxiety medications, or antipsychotics).
The authors concluded that after receiving the results of the florbetapir scan, physicians made significant changes in their diagnoses and had increased diagnostic confidence. They also showed that treatment plans were modified after florbetapir imaging both for patients who were in the midst of their workup and for those with a complete workup.
Cross-sectional study
Landau S, Mintun MD, Joshi A, Koeppe R, Petersen R, Aisen P, et al. Amyloid Deposition, Hypometabolism, and Longitudinal Cognitive Decline. Ann Neurol 2012;72:578–586.
Landau and associates performed a study using longitudinal multisite data to examine the cross-sectional relationships between amyloid deposition, hypometabolism, and cognition, and the associations between amyloid and hypometabolism measurements, and retrospective, longitudinal cognitive measurements. In this study, 426 Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with an available florbetapir and MRI scan were enrolled (126 normal, 162 early mild cognitive impairment (EMCI), 85 late mild cognitive impairment (LMCI), 53 Alzheimer’s disease (AD); 417 of these participants also had an FDG-PET scan acquired approximately concurrently with the florbetapir scan (average time between FDG-PET and florbetapir, < one week). Approximately 2/3 of the total sample were newly enrolled subjects who had no longitudinal follow-up, whereas approximately 1/3 were continuing normal (n = 76) and LMCI (n = 81) participants from ADNI 1 who were followed for an average of about four years prior to their florbetapir scans.
Inclusion as well as exclusion criteria were specified and followed. LMCI participants had the following characteristics: a subjective memory complaint, a Clinical Dementia Rating (CDR) of 0.5, and were classified as single- or multi-domain amnestic. The EMCI group differed from LMCI group only based on education-adjusted scores for the delayed paragraph recall sub-score on the Wechsler Memory Scale–Revised Logical Memory II, such that EMCI subjects were intermediate between normal subjects and LMCI. Normal subjects had CDR scores of 0, and patients with AD met standard diagnostic criteria. The ADAS-cog16 was used in the cross-sectional analyses and well as the primary outcome measure in the longitudinal analyses (total score ranges from 0 to 70, with a higher score indicating poorer cognitive function). Changes in diagnostic status (e.g., remaining LMCI or converting to AD) were also assessed. In the study, ApoE genotypes were determined with blood samples in all except two EMCI subjects. PET image data were acquired based on ADNI protocol. The associations between concurrent florbetapir, FDG, and ADAS-cog measurements for the whole population and for each diagnostic group separately (normal, EMCI, LMCI, AD) were obtained; Spearman rank correlation coefficients were used for continuous variables to account for the non-normally distributed nature of florbetapir and ADAS-cog, and chi-square tests were used for dichotomous variables. For participants with longitudinal data, associations between independent variables (florbetapir and FDG PETs) and longitudinal ADAS-cog change were explored using linear mixed effects models.
Results of the study revealed that 29% of normal subjects, 43% of EMCI patients, 62% of LMCI patients, and 77% of AD patients were categorized as florbetapir positive, and florbetapir was negatively associated with concurrent FDG and ADAS-cog in both MCI groups. The longitudinal analysis also revealed that florbetapir-positive subjects in both normal and LMCI groups had greater ongoing ADAS-cog decline than those who were florbetapir negative, though in normal subjects, florbetapir positivity was associated with greater ADAS-cog decline than FDG, whereas in LMCI, FDG positivity was associated with greater decline than florbetapir.
The authors concluded that, although both hypometabolism and Aβ deposition were detectable in normal subjects and all diagnostic groups, Aβ showed greater associations with cognitive decline in normal participants. In view of the minimal cognitive deterioration overall in this group, the authors felt that the study suggested that amyloid deposition has an early and subclinical impact on cognition that might precede metabolic changes. They also concluded that at moderate and later stages of disease (LMCI/AD), hypometabolism becomes more prominent and more closely linked to cognitive decline.
Additional Studies submitted during the Second Comment Period - (July 3, 2013 – August 2, 2013)
Johnson KA, Sperling RA, Gidicsin RA, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimer’s & Dementia. 30 January 2012:1-12.
Johnson and associates used florbetapir to perform a study to assess amyloid burden, using visual as well as quantitative measures (Johnson, Sperling, Gidicsin, et. al 2012). This multi-center, phase II investigation included 45 patients with AD, 60 patients with MCI, and 45 apparently normal healthy patients in the control group. Results of the study revealed that florbetapir PET imaging was rated visually amyloid positive in 76% of AD patients, 38% of MCI patients, and 14% of HCs. Also 84% of AD patients, 45% of MCI patients, and 23% of HCs were classified as amyloid positive using the quantitative threshold. It also revealed that amyloid positivity and mean cortical amyloid burden were associated with age and apolipoprotein E ε4 carrier status.
The authors acknowledged that the percentage of subjects rated positive, particularly for the AD and MCI groups, was less than in some previous studies using other PET amyloid tracers, and gave several explanations (e.g., the percentage of subjects who were APOE ε4 carriers in the current study (40% of MCI patients and 53% of AD patients) was lower than in previous APOE ε4-enriched multicenter research studies; the selection criteria may have contributed to the lower observed rate of amyloid-positive cases). They also noted that some of the image readers in the study appeared to be more conservative in their interpretation, and potentially less sensitive to the presence of tracer accumulation/amyloid pathology in comparison with the quantitative analysis, and even noted that one reader did show a higher overall rate of positivity than the others. Finally, a post-mortem examination, required for the gold standard diagnosis of AD, was not part of the study.
Zannas AS, Doraiswamy PM, Shpanskaya KS, et al. Impact of 18F-florbetapir PET imaging of β-amyloid neuritic plaque density on clinical decision-making. Neurocase. 14 May 2013:1-8.
Zannas and associates performed a case series study; the objective was to determine if clinical management changed based on the results of florbetapir PET imaging (Zannas et.al 2013). The study involved 11 cognitively impaired subjects. Clinician surveys were done before and after PET scanning to document the impact of amyloid imaging on the diagnosis and treatment plans. All patients had dementia or MCI as a pre PET diagnosis. Of the patients involved in the study, four were felt to have AD as the etiology; the rest were suspected of having depression, vascular disease or another etiology. Results of the study were mixed. It revealed that in five cases, the florbetapir test was negative, leading to a change in diagnosis in four patients, and a change in treatment in two cases. In six cases, the test was positive leading to a change in diagnosis in four patients and a change in treatment plan in three of these cases. But the authors were also able to document cases were patients were suspected of having MCI or depression, and even though their test were positive for florbetapir, there was no change in management. Also the authors noted a case of an MCI patient that was kept on cholinesterase inhibitors treatment despite a negative test. None of the patients were followed longitudinally long enough in order to have a post mortem examination of the brain—the gold standard for the diagnosis of AD.
Choi SR, Scheider JA, Bennett BA, et al. Correlation of amyloid PET ligand florbetapir F 18 (18F-AV-45) binding with β-amyloid aggregation and neuritic plaque deposition in postmortem brain tissue. Alzheimer Disease and Associated Disorders. 2012 January;26(1):8–16.
Choi and associates studied the ability of florbetapir F 18 to accurately identify and quantify amyloid aggregates in human autopsy brain tissue (Choi et. al. 2013). The purpose of their study was to determine the relationship between florbetapir F 18 tissue retention as measured by autoradiography (ARG) and the localization of amyloid plaques using double-labeling studies. They also wanted to determine the correlation between the intensity of the florbetapir ligand signal and β-amyloid deposition. In the study the postmortem brain tissue of 40 subjects suffering with varying degrees of neurodegenerative pathology was assessed using florbetapir F 18 autoradiography (subjects chosen to represent a range of pathologic diagnoses including subjects free of pathology, subjects with AD, subjects with vascular dementia and subjects with progressive supranuclear palsy), and later correlated with β-amyloid identified utilizing silver staining, thioflavin S staining, and immunohistochemistry.
The study was able to demonstrate that there was a strong correlation between the density of in vitro florbetapir F 18 binding in human autopsy tissue, and that there was a strong correlation between the density of in vitro florbetapir F 18 binding and the density of β-amyloid. The authors also noted that the intensity of the florbetapir F 18 signal in human autopsy sections was correlated with the degree of ligand binding in regional brain homogenates; and that florbetapir F 18 does not bind to neurofibrillary tangles in human postmortem tissue.
Though the authors concluded that florbetapir F 18 can be used as an amyloid PET ligand to identify the presence of AD pathology in patients with signs and symptoms of progressive late-life cognitive impairment, they provided little information on the degree of correlation of florbetapir F 18 in patients with conditions other than AD (e.g., subjects free of pathology, subjects with vascular dementia and subjects with progressive supranuclear palsy).
4. MEDCAC
A Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) meeting was convened on the role of PET Aβ imaging in dementia and neurodegenerative disease on January 30, 2013. The purpose was to seek the expert panel’s input on whether the published evidence identified patient characteristics that would predict improved health outcomes for patients who undergo PET Aβ imaging. The panel voted on a series of questions using a 1-5 confidence scale (with 1 representing low or no confidence; 3, intermediate confidence; and 5, high confidence).
A key question for the panel was: How confident are you that there is adequate evidence to determine whether PET imaging of brain beta amyloid changes health outcomes (improved, equivalent or worsened) in patients who display early symptoms or signs of cognitive dysfunction? The average score of voting panel members was below an intermediate level (2.17 out of 5).
The record of the MEDCAC meeting is available on the CMS website. We hereby incorporate it into the administrative record of this NCD by reference. (http://www.cms.gov/medicare-coverage-database/details/medcac-meeting-details.aspx?MEDCACId=66).
5. Evidence-based guidelines
We searched the National Guideline Clearinghouse (www.guidelines.gov) and the Internet more generally for relevant guidelines.
Keith A. Johnson, Satoshi Minoshimab, Nicolaas I. Bohnen, Kevin J. Donohoe, Norman L. Foster, Peter Herscovitch, Jason H. Karlawish, Christopher C. Rowe, Maria C. Carrillo, Dean M. Hartley, Saima Hedrick, Virginia Pappas, William H. Thies. Appropriate use criteria for amyloid PET: A report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. First published January 28, 2013, doi: 10.2967/jnumed.113.120618 J Nucl Med March 1, 2013 jnumed.113.120618
Given that PET Aβ imaging “is a technology that is becoming more available,” the Amyloid Imaging Taskforce (AIT) formed jointly by the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association, sought “to provide guidance to dementia care practitioners, patients, and caregivers” on its appropriate use.
A summary of the AIT’s appropriate use criteria appears below:
“Amyloid imaging is appropriate in the situations listed here for individuals with all of the following characteristics: Preamble: (i) a cognitive complaint with objectively confirmed impairment; (ii) AD as a possible diagnosis, but when the diagnosis is uncertain after a comprehensive evaluation by a dementia expert; and (iii) when knowledge of the presence or absence of Aβ pathology is expected to increase diagnostic certainty and alter management.
- Patients with persistent or progressive unexplained MCI
- Patients satisfying core clinical criteria for possible AD because of unclear clinical presentation, either an atypical clinical course or an etiologically mixed presentation
- Patients with progressive dementia and atypically early age of onset (usually defined as 65 years or less in age)
Amyloid imaging is inappropriate in the following situations:
- Patients with core clinical criteria for probable AD with typical age of onset
- To determine dementia severity
- Based solely on a positive family history of dementia or presence of ApoE4
- Patients with a cognitive complaint that is unconfirmed on clinical examination
- In lieu of genotyping for suspected autosomal mutation carriers
- In asymptomatic individuals
- Nonmedical use (e.g., legal, insurance coverage, or employment screening)”
6. Professional Society Position Statements
A handful of nuclear medicine and physician professional societies, and AD/dementia organizations commented on the PET Aβ proposed decision memo, which we responded to in the Public Comment section below. These comments can be viewed in their entirety at: http://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=265.
7. Expert Opinion
We sought and received expert opinion through the MEDCAC process. We also received expert opinion during our public comment period.
8. Public Comments
A. Initial Comment Period: October 9, 2012 – November 8, 2012
CMS received 27 timely public comments during the first public comment period. Twenty-six out of 27 commenters supported Medicare coverage of PET Aβ scans in the diagnostic context of suspected dementia. Of the supporting commenters, a few wrote that Aβ imaging agents should not be covered for screening of asymptomatic patients, patients without documented cognitive decline, or patients whose AD diagnosis could be confirmed without a PET Aβ scan. Another supportive commenter stated that the meaning of a positive or negative PET Aβ scan, as outlined in the FDA-approved label, should be fully communicated by providers to patients.
The non-supportive commenter argued that research on Aβ imaging agents (particularly Amyvid™ (florbetapir), as the only FDA-approved Aβ imaging agent to date) is too limited, and does not demonstrate a beneficial impact on clinical management of dementia and on health outcomes. This commenter did, however, support the use of Amyvid™ in clinical trials.
Comments came from the following sources:
- 1 (4%) comment came from physicians;
- 7 (26%) comments came from the pharmaceutical and PET imaging industry;
- 5 (18%) comments came from medical imaging societies and specialty groups;
- 9 (33%) comments came from researchers or persons at academic institutions;
- 1 (4%) comment came from the health insurance industry;
- 1 (4%) comment came from research hospitals;
- 2 (7%) comments came from Alzheimer’s societies (USAgainstAlzheimer's and Alzheimer’s Foundation of America); and
- 1 (4%) comment came from members the general public who did not identify a further affiliation.
B. Second Comment Period: July 3, 2013 – August 2, 2013
CMS received 202 timely public comments on the proposed decision. Many of the public comments we received cited unpublished evidence such as data presented at conferences and the results of individual practitioners or patients (often on behalf of family members and caregivers). CMS took into consideration all public comments. We respond in detail to major themes in the public comments below.
The public commenters raised eight key concerns. Several commenters:
(1) raised concerns regarding the CMS standard for making a reasonable and necessary determination for diagnostic tests;
(2) believed that CMS should cover amyloid PET to help differentiate frontotemporal dementia (FTD) from AD since CMS has covered FDG PET for this use;
(3) suggested that the final decision should more closely reflect the recommendations by expert consensus panels;
(4) state or imply that a PET amyloid scan gives an accurate, positive diagnosis of AD;
(5) claimed that dementia specialists could make an accurate positive diagnosis of AD when integrating the result of an amyloid PET scan.
(6) suggested that because the FDA approved the amyloid PET agent florbetapir (Amyvid™), CMS should cover a diagnostic test using that agent;
(7) believed that our proposed decision permitting coverage only in certain qualifying clinical studies would be inconsistent with the National Alzheimer’s Project Act (NAPA); and/or
(8) believed the proposed decision would be too onerous and restrictive and would limit access to this new technology.
We address the above concerns in detail in our response to the comments.
CMS standard for making a reasonable and necessary determination for diagnostic tests
Comment
Several commenters believe that evidence of “improved health outcomes” should not be a factor for a coverage determination on amyloid PET.
Response
We disagree. Section 1862(a)(1)(A) of the Act states that no payment may be made for items or services “which are not reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member.” When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether the evidence is of sufficient quality to support a finding that an item or service that falls within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. This critical appraisal of the evidence enables us to determine whether: 1) the specific assessment questions can be answered conclusively; and 2) the investigational item or service will improve health outcomes for patients. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
Specifically with regard to diagnostic tests, the Medicare regulations at 42 CFR § 410.32(a) state in part, that "…diagnostic tests must be ordered by the physician who is treating the beneficiary, that is, the physician who furnishes a consultation or treats a beneficiary for a specific medical problem and who uses the results in the management of the beneficiary’s specific medical problem.” Thus, we looked for evidence demonstrating how the treating physician uses the result of beta amyloid PET imaging for the management of a patient with suspected AD.
In evaluating diagnostic tests, Mol and colleagues (2003) reported: "Whether or not patients are better off from undergoing a diagnostic test will depend on how test information is used to guide subsequent decisions on starting, stopping, or modifying treatment. Consequently, the practical value of a diagnostic test can only be assessed by taking into account subsequent health outcomes." When a proven, well established association or pathway is available, intermediate health outcomes may also be considered. For example, if a particular diagnostic test result can be shown to change patient management and other evidence has demonstrated that those patient management changes improve health outcomes, then those separate sources of evidence may be sufficient to demonstrate positive health outcomes from the diagnostic test.
A diagnostic test would not be expected to directly change health outcomes. Rather, a diagnostic test affects health outcomes through changes in disease management brought about by physician actions taken in response to test results. Such actions may include decisions to treat or withhold treatment, to choose one treatment modality over another, or to choose a different dose or duration of the same treatment. To some extent the usefulness of a test result is constrained by the available treatment options. Unfortunately the data are silent on health outcomes, and do not establish that the treating physicians appropriately base patient management on the PET test result. Most studies have focused on test characteristics and have not considered health outcomes. We believe that health outcomes are more persuasive than test characteristics.
We generally consider the evidence in the hierarchical framework of Fryback and Thornbury (1991) where Level 2 addresses diagnostic accuracy, sensitivity and specificity of the test; Level 3 focuses on whether the information produces change in the physician’s diagnostic thinking; Level 4 concerns the effect on the patient management plan, and Level 5 measures the effect of the diagnostic information on patient outcomes. CMS has generally found evidence of efficacy at Level 5 more persuasive to support unconditional coverage. We believe that coverage supported by that level or higher evidence results in the greatest benefit for Medicare beneficiaries.
The expectation that a diagnostic test will produce relevant information that informs physician management is well established in the practice of medicine and is also reflected in our regulation (42 CFR 410.32). Accordingly, we ask: Does the test lead the physician to reconsider the pre-test treatment plan and make appropriate modifications in light of the test result? Such actions may include decisions to treat or withhold treatment, to choose one treatment modality over another, or to choose a different dose or duration of the same treatment. There is no persuasive evidence that amyloid PET testing produces relevant information for these purposes.
Specifically for amyloid PET, and as discussed in the analysis and discussions sections of this decision memorandum, there is no convincing evidence that the scan changes physician management of the patient in a meaningful manner (e.g., there is no convincing benefit to Medicare beneficiaries). However, we believe there is promising evidence to cover amyloid PET under coverage with evidence development (CED) and that the test has a high potential to provide a significant benefit to Medicare beneficiaries in the future. Per the CED guidance document, when the evidence is inadequate to determine that the item or service is reasonable and necessary under section 1862(a)(1)(A), Medicare coverage may be extended to patients enrolled in a clinical research study. In this case, AHRQ and CMS are supporting research under section 1862(a)(1)(E). For the readers’ convenience, the 2006 CED Guidance Document is available at http://www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/CED.pdf
We believe that beneficiaries would benefit from the use of the amyloid PET scan to enrich clinical trials and help find better treatments or prevention strategies for AD.
CMS covered FDG PET to differentiate frontotemporal dementia (FTD) from AD
Comment
Several commenters claimed that because CMS currently covers FDG PET to help differentiate frontotemporal dementia (FTD) from AD, amyloid PET should also be covered because they believe it is a similar technology for the same diagnosis and that amyloid PET is a better diagnostic tool. They ask that amyloid PET should be similarly covered, without CED.
Response
In 2004 CMS issued an NCD to cover FDG PET scans for either the differential diagnosis of frontotemporal dementia (FTD) and Alzheimer’s disease (AD) under specific requirements; OR, its use in a CMS-approved practical clinical trial focused on the utility of FDG PET in the diagnosis or treatment of dementing neurodegenerative diseases (see NCD manual, section 220.6.13). FDG PET is a fundamentally different – not a similar – technology. FDG PET measures the physiological process of metabolism, while amyloid PET looks at the anatomical burden of amyloid plaques.
The proposed clinical use of the amyloid PET scan to differentiate FTD from AD leverages the power of a negative scan to help exclude AD, which is consistent with the FDA-approved label and our own detailed assessment. However, the evidence for the scan’s possible clinical utility comes from very small, or yet to be published studies. In response to our concern about the small sample sizes, the lead author of one such study wrote in the public comments that he has soon-to-be published data expanding this patient pool from 12 to 25 subjects, with consistent results. It is encouraging to hear that the data will be published and we look forward to reviewing the data. However, we note that 25 subjects is still a very small sample size in light of reports that over five million Americans age 65 and over have AD. (https://www.alz.org/downloads/facts_figures_2012.pdf)
We also note that many of the commenters appear to assume that amyloid PET is a better tool than FDG PET for differentiating FTD from AD. This may or may not be true – the evidence is not clear – and there is at least some evidence that FDG PET is actually better (as another distinguished PET researcher argues in the public comments, citing peer-reviewed publications). While outside the scope of this NCD, we encourage further study, involving prominent researchers on amyloid PET and FDG PET alike, to help build the evidence base, and determine which of multiple potentially useful tests should be used, when alone or in combination, and for which particular subpopulations (recall that FTD has multiple subtypes, and one algorithm may not fit all of them).
The differentiation of FTD from AD may be one clinical use where CED leads to earlier and broader coverage than would otherwise be accomplished. In addition, our goal under CED is to facilitate the development of additional evidence that will assist practitioners and beneficiaries in determining the best management strategy for patients with suspected AD, based on the results of amyloid PET imaging. We are eager to see new and greater published evidence that amyloid PET could help resolve other such narrowly defined and clinically difficult differential diagnoses, where use of the scan may prove to offer tangible benefits to the patient. Health outcomes of interest, again, include, but are not limited to, any of the following: avoiding inappropriate and potentially harmful medications; avoiding futile or burdensome treatments or tests; improving, or slowing the decline of, quality of life; and survival.
Recommendations by expert consensus panels
Comment
Numerous commenters stated we should accept the recommendations of the AIT (Amyloid Imaging Taskforce) consensus panel regarding the appropriate use of amyloid PET.
Response
The persuasiveness of expert opinion is constrained by the available evidence. Depending on the evidence, expert opinion may vary from conjectural to conclusive. While we recognize and respect the expertise of the AIT panelists, we believe that significant questions still remain open, and that CED can help develop the right studies to answer them.
Furthermore, we also recognize the expertise of another relevant expert consensus panel that we convened on January 30, 2013 – the MEDCAC (Medicare Evidence Development & Coverage Advisory Committee). As noted earlier, the MEDCAC proceeding is available on the CMS website and we refer the reader there for a more detailed account. The MEDCAC:
- includes experts not only on the clinical subject at hand, but also on biostatistics, epidemiology and ethics; and it taps experts from various clinical disciplines – cardiology, surgery, internal medicine – to broaden perspectives on, and experience with, evidence development more generally;
- is not sponsored by industry or any particular organization; and
- includes external expert speakers who provide transparent and critical views during deliberations.
- conducts its deliberations in a public forum.
A key question for the MEDCAC panel was: How confident are you that there is adequate evidence to determine whether amyloid PET imaging of brain beta amyloid changes health outcomes (improved, equivalent or worsened) in patients who display early symptoms or signs of cognitive dysfunction? The mean score of voting panel members was 2.17 (on a scale of 1 to 5, where 1 represents “low confidence,” 5 represents “high confidence,” and 3 represents “intermediate confidence”).
Although the MEDCAC did not find sufficient evidence for CMS to support outright coverage of amyloid PET, this comment by one guest panel member – “coverage with evidence development would help fill in a lot of very substantial questions” – echoed comments by multiple panel members (See part 00279 lines 10 – 20 of the MEDCAC transcript available at http://www.cms.gov/Regulations-and-Guidance/Guidance/FACA/Downloads/id66d.pdf). We note that the MEDCAC panel does not actually vote on whether they think CMS should pursue CED.
The MEDCAC also considered the recommendations of the AIT and others during its deliberations. These two credible expert panels – the AIT and the MEDCAC – produced differing consensuses. This highlights the limitation of consensus panels: if you change panel members, you might well change the consensus. That’s why, in the well-established process of scientific evaluation, evidence must be evaluated to determine the strength of the consensus opinion (see Appendix A).
As for the AIT, we acknowledge the difficulty in crafting recommendations in light of the limitations of the currently available evidence. We have recognized numerous points the AIT makes and have included those in this decision where appropriate. This includes the AIT July 2013 update that dementia specialists are better equipped to order such scans than other types of physicians.
We continue to believe based on our review of the published, peer-reviewed medical literature that the evidence gaps for amyloid PET, and AD biomarkers generally, as noted in the AIT’s publication as well as in the 2011 NIA-AA series of guidelines, are consistent with the current CMS decision for CED (see our discussion of biomarkers in the Background and Analysis sections). For example, the AIT does not identify objectively-defined subpopulations of patients with cognitive impairment for which the scan (alone or combined with other tests) may be more or less appropriate. Yet there are many subtypes of MCI, and some (e.g., amnestic MCI) may be more relevant than others. Furthermore, there is evidence that the same level of amyloid burden detected by a scan may mean something very different in say, a 66 year-old compared to an 86 year-old (e.g., Le Couteur 2013, Laforce 2011). Yet the AIT is silent about such potentially important distinctions.
Widespread clinical use of the scan both in many types of patients with unexplained MCI, and to make a positive diagnosis of AD (despite insufficient evidence on the clinical meaning of a positive scan) has great potential to lead to over-diagnosis of AD. Such misdiagnosis of AD portends real harm to our beneficiaries (La Couteur 2013), and this must be considered in our coverage decision. Therefore, we believe CED is appropriate to encourage more studies that will benefit Medicare beneficiaries by answering some of these outstanding questions.
Diagnosis of Alzheimer's disease
Comment
Numerous commenters state or imply that an amyloid PET scan gives an accurate, positive diagnosis of Alzheimer’s disease. The commenters further claim that such use is consistent with the FDA-approved labeling.
Response
We disagree with the commenters. An amyloid PET scan does not give an accurate positive diagnosis of AD, and a claim that it does is inconsistent with the FDA-approved label. Moreover, the FDA Medical Review of florbetapir PET notes that there are two pathophysiological hallmarks of AD which contribute to the gold-standard diagnosis – neurofibrillary tangles of the protein tau, and neuritic amyloid plaques and the amyloid PET scan detects only one of these. Finally, the scan does not distinguish between diffuse and neuritic amyloid plaques, and the significance of this lack of distinction remains unclear.
The positive diagnosis of AD requires not only both of these pathophysiological hallmarks, but also clinical documentation of progressive dementia, and exclusion of other diseases as the cause of the dementia. Because presence of neuritic amyloid plaques is one of the requirements for diagnosing AD, exclusion of the same excludes that diagnosis. Accordingly, the FDA-approved label states that a negative scan “is inconsistent with” a diagnosis of AD.
However, the presence of additional elements are required for the diagnosis of AD, and it is not clear that a certain threshold of amyloid definitively predicts these other elements. The FDA-approved label for Amyvid™ does not make any similar statement on the meaning of a positive scan. Moreover, the FDA notes that similar amyloid levels “may also be present in patients with other types of neurologic conditions as well as older people with normal cognition.” In other words, a positive scan is not necessarily consistent with a diagnosis of AD. This conclusion is consistent with the 2011 NIA-AA consensus guidelines which state that although the presence of amyloid plaques is “necessary,” it is not necessarily “sufficient,” for diagnosing AD. More importantly, this conclusion – that the meaning of a positive scan is unclear – is consistent with the evidence that appears in published clinical studies discussed in the Evidence section.
Integrating the amyloid PET scan in diagnosing AD
Comment
Many commenters claim that dementia specialists could make an accurate positive diagnosis of AD when integrating the result of an amyloid PET scan.
Response
Prior to the publication of our proposed decision memorandum (PDM), the industry-sponsored Grundman (2012) study was the sole prospective study exploring the impact of scan results on physicians’ diagnosis of AD, as well as their subsequent intended clinical management. The Grundman study design assumes that physicians can use the scan to make an accurate diagnosis, but does not demonstrate that they can (as there is no reference to a gold standard diagnosis of AD in the study); nor do any prior research studies demonstrate this.
Studies prior to Grundman 2012 do not report predictive values of the test for AD. The published data are limited to sensitivity (Sn) and specificity (Sp) values for the detection of amyloid alone. Yet these (Sn and Sp) are not the most clinically meaningful values for a diagnostic test. In the case of amyloid PET, while a “negative” test appears to minimize the risk of AD, the meaning of a “positive” test for any particular individual with unexplained cognitive impairment is unclear, and this again could lead to over-diagnosis of AD.
Positive and negative predictive values for AD are more useful than Sn and Sp – they can tell you the meaning of scan results for particular patients who belong to risk-stratified populations – but unfortunately no currently available study presents data on these values. Predictive values for AD mathematically include not only computations for Sn and Sp, but also the quantitative prevalence of disease in objectively-defined patient subpopulations or “risk pools.” But the risk pools for AD are themselves not yet even defined in the literature. Because predictive values corresponding to a “positive” or “negative” test result vary, depending on the “risk pool” the patient objectively falls into, test results absent such values have no clinical meaning for an individual patient.
Since the publication of our PDM, studies similar to Grundman (e.g. Zannas 2013) have emerged, but present similar limitations and thus inclusive results. (Please see further discussion in Section VIII: CMS Analysis, below.)
FDA approval and CMS coverage.
Comment
Several commenters stated that CMS should automatically cover amyloid PET scans because the FDA approved the PET amyloid agent florbetapir (Amyvid™).
Response
FDA premarket review and CMS national coverage determinations differ significantly. Each process operates under different statutory standards and each asks different questions to meet its respective mandates. The FDA premarket review generally assesses the safety and effectiveness of these medical products. Even within FDA's review processes, there are differences in types of evaluation depending upon the application under consideration (for example, premarket approval applications (PMAs) must meet standards different from premarket clearance (510(k)).
CMS serves a different function by providing health insurance to protect the nation's aged and disabled persons from the substantial burdens of illness. Under section 1862(a)(1) of the Act, CMS makes determinations regarding the coverage of specific items and services. In short, CMS must make multiple decisions: It must decide what items and services it can and should pay for; how it should accomplish the payment; and how much to pay.
CMS' evaluation of medical products depends on the type of request. For most NCDs, CMS evaluates whether a medical product or service is reasonable and necessary to diagnose or treat an illness or injury affecting the Medicare population. This evaluation includes review of appropriate outcomes data, such as whether the product provides improved, equivalent, or complementary health outcomes in the Medicare population as compared to alternative treatments or diagnostics already covered by the program. CMS may also evaluate medical product indications that have not been approved or cleared by FDA, so-called unapproved or off-label uses as found in 75 FR 57045, pages 57045 -57048 available at http://www.gpo.gov/fdsys/pkg/FR-2010-09-17/pdf/2010-23252.pdf
In the case of amyloid PET, FDA limited its evaluation essentially to the safety and efficacy of the radiopharmaceutical agent itself – florbetapir (Amyvid™) – that is used in the diagnostic imaging test. We discussed in previous responses to comments CMS statutory and regulatory authority for reviewing items and services for the purposes of Medicare coverage.
National Alzheimer’s Plan Act (NAPA)
Comment
Some commenters claimed that CMS’ proposed decision to cover amyloid PET under CED is inconsistent with NAPA. The commenters state that NAPA supports the coverage of amyloid PET scan as a diagnostic tool for AD.
Response
We believe covering amyloid PET scans under CED supports NAPA. In fact, NAPA’s Strategy 2.B, “Ensure Timely and Accurate Diagnosis,” was intended in part to further the NIH-CMS work on early detection using “assessment tools that can be used to detect cognitive impairment.”
NAPA seeks the development of tools that can help ensure an “accurate diagnosis” of AD, but this particular test – amyloid PET – has not been demonstrated to do this. This finding is based on our independent evidence review, and is consistent with the FDA-approved label and supporting FDA Medical Review of florbetapir, as well as technology assessments by other scientific bodies (TEC 2013, EMA 2013). We believe supporting amyloid PET under CED is the best decision for beneficiaries and practitioners. It is our belief that if the appropriate CED trials are completed CED will give information on where this new technology will be most useful in the diagnosis and treatment of AD. This NCD is consistent with and supportive of the NAPA goals in the following ways:
CED supports NAPA’s strategy 1.B “Expand Research Aimed at Preventing and Treating Alzheimer’s Disease.” By CMS requiring CED for the coverage of amyloid PET scans we support any study that meets the criteria outlined in section I. As stated previously, we believe CED is necessary to ensure that beneficiaries are receiving the best care. Based on our review of the evidence, including MEDCAC input, we believe that amyloid PET will be available to Medicare beneficiaries in the context of clinical studies. It is CMS’ belief that these studies are necessary to determine the best use of this diagnostic test.
This decision is consistent with NAPA’s Strategy 1.C, “Accelerate Efforts to Identify Early and Presymptomatic Stages of Alzheimer’s Disease.” As discussed in the analysis and discussion section of this decision memorandum, CMS-approved studies done under CED should help better define subpopulations at risk for developing AD. This is an important question not only for this Medicare population and amyloid PET, but aligns with other ongoing research efforts (e.g. the large, multicenter, NIH-funded Alzheimer’s Disease Neuroimaging Initiative).
Coverage with evidence development (CED)
Comment
Several commenters expressed concern that CED requirements would be too onerous and restrictive. Comments believed that CMS would only approve a single study. Some commenters also stated that no study could answer all of the questions posed in the proposed decision.
Response
We do not believe CED has to be onerous or unduly restrictive. There appear to be many misperceptions about how CED for amyloid PET could be designed. Under this NCD, there are potentially many studies that could meet the CED study criteria outlined in section I of this decision memorandum. CMS is not limited to approving only a single study; any number of studies can be approved as long as the study meets the NCD criteria. Further, a study does not have to attempt to answer all CED questions asked in this NCD, but could focus on any aspect of one or more of the questions (which appear in the Section I: Final Decision).
We stated that these studies should be prospective, randomized, and have autopsy as an endpoint, only when appropriate. The specific clinical study protocol is determined by the nature of the question being asked, and the likely sources of bias and confounding, and we will evaluate the protocols as they are submitted to determine which CED studies appropriately meet the criteria specified in the NCD. In addition, an approved study that meets the NCD criteria might synergize with, or piggy-back on, existing research efforts. Studies might be integrated, involving enrollment in companion or parallel studies. And they might employ newer methods such as “adaptive” or “pragmatic” clinical trial designs.
Comment
Some commenters asked whether a study approved under this NCD could use newer analytical methods to churn on large amounts of cohesive clinical data gathered from use of the scan in “real patients” in “real clinical settings."
Response
We think it is possible. This would be consistent with the vision for the future of research articulated in chapter 6 of the Institute of Medicine’s recent report, “Best Care at Lower Cost: The Path to Continuously Learning Health Care in America” (IOM 2012). We recognize that such data could help not only to close basic evidence gaps, but also to establish “generalizability” of the technology – evidence that beneficial outcomes could be sustained outside the clinical trial setting – as access rolls out to potentially hundreds of thousands of patients (as has actually happened in a prior CED). We encourage submission of clinical research designs that incorporate this vision.
Additional Evidence
Comment
Some commenters provided additional evidence that was not included in the bibliography of the proposed decision memo as sufficient for coverage of amyloid PET in dementia and neurodegenerative disease without CED.
Response
We appreciate the additional references provided in the public comments. We found the three published clinical trials on florbetapir relevant to this NCA and referenced them in the Evidence Section above and the Analysis Section.
Full text public comments without PHI can be viewed at http://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=265
VIII. CMS Analysis
National coverage determinations (NCDs) are determinations by the Secretary of Health and Human Services (“the Secretary”) of whether a particular item or service is covered nationally by Medicare, under §1869(f)(1)(B) of the Act.
In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, §1862(a)(1) of the Act in part states that, with limited exceptions, no payment may be made under Part A or part B for any expenses incurred for items or services:
- which are not reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member (§1862(a)(1)(A)) of the Act; or
- in the case of research conducted pursuant to section 1142, which is not reasonable and necessary to carry out the purposes of that section (§1862(a)(1)(E)) of the Act.
Section 1142 of the Act describes the authority of the AHRQ. Under section 1142, research may be conducted and supported on the outcomes, effectiveness, and appropriateness of health care services and procedures to identify the manner in which diseases, disorders, and other health conditions can be prevented, diagnosed, treated, and managed clinically.
Section 1862(a)(1)(E) of the Act allows Medicare to cover under CED certain items or services where additional data gathered in the context of clinical care would further clarify the impact of these items and services on the health of Medicare beneficiaries. The 2006 CED guidance document is available at www.cms.gov/Medicare/Coverage/DeterminationProcess/downloads/ced.pdf.
Questions:
- Is the evidence adequate to conclude that PET Aβ imaging improves meaningful health outcomes in beneficiaries who display signs and symptoms of AD?
- Is the evidence adequate to conclude that PET Aβ imaging results inform the treating physician's management of the beneficiary to improve meaningful health outcomes? Those outcomes may include reasonably considered beneficial therapeutic management or the avoidance of unnecessary, burdensome interventions.
In the following pages we note the limitations of specific published studies and ultimately our overall conclusions about the body of evidence.
Prospective Longitudinal Studies
Wong D, Rosenberg P, Zhou Y, Kumar A, Raymont V, Ravert H, et al. In Vivo Imaging of Amyloid Deposition in Alzheimer’s Disease using the Novel Radioligand [18F]AV-45 (Florbetapir F18). J Nucl Med. 2010 Jun; 51(6): 913–920.
Wong and associates performed a prospective, open-label, multicenter, brain imaging study to test the pharmacokinetics of the tracer florbetapir and its safety for patients. They concluded that florbetapir PET imaging could discriminate between AD patients and healthy control subjects. But as noted by the authors, there were a number of limitations of the study. The study was small, and 6 of 32 (19%) of planned subjects were not included in the primary analysis due to technical failures during the scanning process. There was limited evaluation of imaging protocols and test efficacy. Also, due to the open-label study design, interpreters could have been biased in reporting results as they were not blinded. Despite its limitations, this study was a stepping stone to efficacy studies (e.g., Clark 2011 and 2012), which used autopsy, not clinical diagnosis, as the gold standard.
Camus V, Payoux P, Barré L, Desgranges B, Voisin T, Tauber C, et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur J Nucl Med Mol Imaging. 2012 Apr;39(4):621-31. doi: 10.1007/s00259-011-2021-8. Epub 2012 Jan 18.
Camus and associates performed a prospective study and concluded that florbetapir PET was “a safe and suitable biomarker for AD that can be used routinely in a clinical environment.” A number of limitations were noted by the authors, including a small sample size (n = 46), and selection bias due to the significantly older age in the MCI group than in the AD and healthy control groups. The authors were also concerned about the short half-day training sessions as well as the low specificity of the visual PET scan assessment, which could result in a high false positive rate, but suggested ways to improve these, such as improving and lengthening the duration of training, increasing the spatial resolution of tomographs, and adopting semiautomatic or automatic quantification methods or software. Finally, clinical diagnosis was used as the reference standard in this study, instead of the postmortem gold standard as used in other studies (Clark 2011, Clark 2012).
Clark CM, Sneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA. Use of Florbetapir PET for Imaging Aβ Pathology. JAMA 2011 Jan 19;305(3):275-83.
The 2011 study by Clark and associates concluded that overall Aβ burden assessed in vivo with florbetapir PET imaging correlates with histopathological assessments at autopsy. The authors acknowledged a number of limitations of the study. First, the sample size of the autopsy cohort was small (n = 35, of which six subjects were used to validate the protocol). Second, the non-autopsy cohort, used to determine the likelihood that a florbetapir PET image could falsely suggest the presence of amyloid, consisted of young, cognitively normal subjects – a distinctly different population from the end-of-life autopsy cohort.
Another limitation of the study was that amyloid scans were interpreted by three trained nuclear medicine physicians and the median of the three results was used in the analysis. The authors acknowledgement that this was “...a process not likely to be replicated in clinical settings” highlights the issue of external validity and the study’s generalizability to the community setting. There was intentional selection bias as subjects chosen were those most likely to provide the shortest possible interval between imaging and histopathological evaluation (e.g., they were likely to die soon). Also, there were no standardized criteria for determining AD or MCI. An additional limitation not stated by the authors is that the use of a semi-quantitative categorical (0 - 4) ranking of florbetapir images, rather than a binary interpretation, limited evaluation of sensitivity and specificity.
Clark C, Pontecorvo M, Bench T, Bedell B, Coleman R, Doraiswamy P. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic Aβ plaques: a prospective cohort study. Lancet Neural 2012;11:669-78.
In the Clark 2011 study, 35 patients had postmortem exams. To this group an additional 24 new subjects with postmortem exam were added for the Clark 2012 study, yielding a total of 59 subjects, whose cognitive status during life ranged from normal to advanced dementia. The authors concluded that florbetapir PET could be used to distinguish patients with no or sparse amyloid plaques from those with moderate to frequent plaques.
Unlike in the 2011 study, all subjects in the 2012 study were end-of-life and underwent a postmortem examination, thus eliminating age cohort as a limitation. Although this issue was addressed, the authors noted several other limitations of the 2012 study. Subjects represented an end-of-life population that is generally older and sicker than those who would seek diagnosis for cognitive impairment in a community setting.
Also, the Clark 2011 study used the median interpretation of three trained nuclear medicine readers, while the Clark 2012 study used the majority interpretation of five trained nuclear medicine readers. This discrepancy (the change in measurement) is a potential violation of internal validity.
Another limitation pointed out by the authors was that both imaging and histopathological results were distributed bimodally, with few “borderline” cases. This raises the question of whether a lower sensitivity might have been obtained if more participants who had intermediate results had been involved. The authors suggested that additional studies would be needed to assess the frequency of such borderline scans, and their implications for performance characteristics of the test, in community settings and with more typical patients. Finally, the authors noted that the “clinical significance of amyloid burden as measured with florbetapir PET must be interpreted in the context of other relevant diagnostic information.”
Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N. Using Positron Emission Tomography and Florbetapir F 18 to Image Cortical Amyloid in Patients With Mild Cognitive Impairment or Dementia Due to Alzheimer Disease. Arch Neurol. 2011;68(11):1404-1411.
Fleischer and associates felt that their study demonstrated that florbetapir PET SUVRs were able to characterize Aβ levels in clinically probable AD, MCI, and older health control groups using continuous and binary measures of fibrillar Aβ burden. But the authors commented on a number of limitations of the study. First, they noted that although mean cortical SUVRs were higher in ApoE4 carriers compared with non-carriers, the proportion of florbetapir PET positivity between carriers and non-carriers did not reach statistical significance. They felt that the small sample size of ApoE4 carriers was probably the reason. Second, there were a lack of standardization for image acquisition, cerebral and reference ROIs, and cut-off thresholds. Third, there was cohort selection bias. Additionally, we note that this study does not use the postmortem gold standard for diagnosing AD; rather, SUVR data from the scans (with a certain cut-off value derived from a small sample in a prior autopsy study) are compared to presence of AD as diagnosed clinically.
Doraiswamy P, Sperling R, Coleman R, Johnson K, Reiman E, Davis, M. Amyloid-β_ assessed by florbetapir F 18 PET and 18-month cognitive decline: A multicenter study. Neurology 2012;79:1636–1644.
The goal of the study performed by Doraiswamy and associates was to evaluate the prognostic use of detecting Aβ pathology using florbetapir PET in subjects at risk for progressive cognitive decline. The authors concluded that florbetapir PET may help identify individuals at increased risk for progressive cognitive decline, but identified a number of limitations of the study. They noted that the lower-than-expected conversion rates among the Aβ positive patients (compared to prior PIB studies) could have been due to the low sample size as well as the short duration of the study. They also noted that subjects with MCI in this study were less impaired at baseline compared to subjects with MCI in the Alzheimer’s Disease Neuroimaging Initiative (ADNI; another study assessing neuroimaging in patients with AD). This was felt likely due to differing entry criteria as well as selection bias. This study did not collect other biomarker data (e.g., ApoE4) and could not assess the relative utility of PET versus other biomarkers. Also, the reference standard for AD was clinical diagnosis, not the postmortem gold standard.
In this study a positive scan was determined by the majority read of three nuclear medicine physicians. As has been noted before, this may not be replicated in clinical settings. Finally, the authors believe that larger, “longitudinal PET and cognitive data may help clarify its prognostic role in the clinical setting, its ability to improve [diagnostic] confidence . . . and for subject enrichment of therapeutic trials in the early clinical and preclinical stages of AD.”
Grundman M. Pontecorvo M, Salloway S, Doraiswamy P, Fleisher A, Sadowsky C, et al. Potential Impact of Amyloid Imaging on Diagnosis and Intended Management in Patients With Progressive Cognitive Decline. Alzheimer Dis Assoc Disord 2012;00:000–000.
Grundman and associates sought to demonstrate that the use of florbetapir PET scans altered self-reported physician diagnosis and increased their diagnostic confidence. The researchers felt that the study showed that treatment plans were modified after florbetapir imaging both for patients who were in the midst of their workup and for those with a complete workup. But the study had a number of limitations, many noted by the authors. First, the study recorded intended change in management, but it did not evaluate actual change in management. Second, there was intentional selection bias. Patients were subjectively selected for “specific attributes,” and while they likely overlap populations of diagnostic interest, these populations were not defined, limiting the study’s generalizability. Third, no postmortem gold standard was used. Finally, because expert nuclear medicine specialists over-read the scans, and the study was carried out in a clinical trial setting, where participating physicians were largely experts experienced in the diagnosis and/or care of AD patients, it may be difficult to duplicate the study’s findings in a general setting.
Landau S, Mintun MD, Joshi A, Koeppe R, Petersen R, Aisen P, et al. Amyloid Deposition, Hypometabolism, and Longitudinal Cognitive Decline. Ann Neurol 2012;72:578–586.
Landau and associates concluded that a positive PET Aβ test in both the normal and late MCI patients (LMCI) groups was associated with ongoing decline, though in normal subjects, decline was more closely linked to amyloid status, whereas in LMCI, decline was more closely linked to hypometabolism. The researchers also acknowledge some limitations of the study. First, the associations with longitudinal cognitive decline are retrospective rather than predictive, as the florbetapir and FDG measurements were collected at the end of the follow-up period. Second, the distributions of FDG PET and florbetapir differ: florbetapir was more bimodal than FDG PET. Thus the use of dichotomous predictor variables may more accurately reflect the underlying characteristics of the florbetapir distribution. Additionally, we note that the reference standard for AD was clinical diagnosis, not the postmortem gold standard. Finally, cross-sectional data was used to show the relationships between Aβ (measured with florbetapir), hypometabolism (measured with FDG PET), and cognitive performance – and such cross-sectional designs are prone to ecological fallacy.
Three additional studies were submitted during the second comment period (Johnson et al. 2012, Zannas et al. 2013, Choi et al. 2012). Though Johnson and colleagues were able to demonstrate a lower frequency of amyloid burden as they compared AD patients to MCI patient and healthy controls, their results were consistently lower when compared to previous studies using other PET amyloid tracers. The authors listed a number of possible explanations including selection criteria as well as image reader variability. These factors could negate the findings of the study, thus calling into question its validity.
Zannas and associates’ objective was to determine if clinical management changed based on the results of the florbetapir PET image. Though this was a small case series study, when results were obtained, it revealed that there were inconsistencies in management based on results: some patients who tested negative for beta amyloid were kept on Alzheimer’s medications, while some patients who were thought to have depression or MCI who tested positive for beta amyloid plaque, were never treated with Alzheimer’s medications.
Choi and colleagues evaluated the ability of florbetapir F18 to identify and quantify amyloid aggregates in autopsied brain tissue. Their study involved the use of brain specimens taken from a range of patients including those with AD, vascular dementia, progressive supranuclear palsy, and normal subjects. Though they were able to demonstrate a strong correlation between the density of invitro florbetapir F18 in patients with late-life cognitive impairment, they provided little information on the degree of correlation in patients with conditions other than AD.
While the addition of these articles served to round out the currently available evidence base for beta amyloid PET imaging in dementia and neurodegenerative disease, it did not change our final decision. The evidence is insufficient to conclude that beta amyloid PET is reasonable and necessary; it is sufficient for coverage under CED.
A. Discussion
The clinical usefulness of AD testing, including PET Aβ imaging, is limited by the current absence of therapies that meaningfully prevent, stabilize or reverse the progressive course of the condition. This leads to a corresponding limitation in the evidence that might be brought to bear on the impact of testing on meaningful clinical outcomes. Thus we have no evidence that PET Aβ imaging leads through informed physician management to the prevention, stabilization or reversal of AD.
That said, we recognize that there are other incurable conditions, for example, some cancers, where the prudent use of diagnostic testing can meaningfully inform physician decision-making and patient management. In the case of cancer, a positive imaging test that leads to a definitive diagnosis by biopsy could reasonably guide physician management toward palliative goals that are acceptable to the patient and consistent with scientific evidence. Thus we are open to reasoned, evidence-based arguments that would identify benefit that may be achieved by the avoidance of burdensome or hazardous interventions that will not ultimately help the beneficiary.
The expectation that a medical test inform physician management is well established. It is also consistent with federal regulation at 42 C.F.R. §410.32(a), which requires that:
“. . . diagnostic tests must be ordered by the physician who. . . treats a beneficiary for a specific medical problem and who uses the results in the management of the beneficiary’s specific medical problem.”
Accordingly, we ask: Does the test lead the physician to reconsider the pre-test treatment plan and make appropriate modifications in light of the test result? What evidence is available to support assertions of benefit from testing?
We recognize that the medical literature often describes test characteristics and has not consistently considered the impact of testing on physician decision making and patient health outcomes, such as mortality, morbidity or reduction of invasive testing. However, we believe that evidence of improved health outcomes is more persuasive than descriptions of test characteristics. (Please see Appendix A: General Methodological Principles of Study Design).
In evaluating diagnostic tests, Mol (2003) states: “Whether or not patients are better off from undergoing a diagnostic test will depend on how test information is used to guide subsequent decisions on starting, stopping or modifying treatment. Consequently, the practical value of a diagnostic test can only be assessed by taking into account subsequent health outcomes.” For example, we recognize that if a particular diagnostic test result can be shown to change patient management, and if other evidence has confidently demonstrated that those patient management changes improve health outcomes, then a combination of such sources of evidence may be sufficient to demonstrate positive health outcomes from the diagnostic test. We also note for completeness that we are unaware of any claims that florbetapir administration itself exerts any direct therapeutic effect.
Prior to the posting of the proposed decision memo, the industry-sponsored Grundman (2012) study was the sole prospective study attempting to measure the impact of scan results on intended clinical decision making and management. Since that time, other studies (e.g., Zannas 2013) have tried to explore this relationship but have been met with mixed results (see above response to public comments).
The Grundman 2012 authors opine (and we agree), that “[a] remaining question is whether clinical care that includes amyloid imaging will translate into better outcomes.” Grundman also states that “[a]dditional longitudinal studies would be required, however, to explicitly quantify the relationship between amyloid imaging and patient outcomes.” Our overall assessment of Grundman 2012 is that it is a good hypothesis-generating study. It raises the possibility that PET Aβ scans could improve medication management, and reduce other testing, but does not establish these conclusively. Also, its lack of objective criteria, both for patient selection and for changes in decision-making and management, markedly limit its ability to inform community practice outside of a clinical study. We now discuss this assessment in more detail.
We mentioned earlier that virtually all subjects in Grundman 2012 who had a positive amyloid scan ended up being given a clinical diagnosis of AD by the physicians (112/113), while virtually all patients who had a negative amyloid scan ended up being given a clinical diagnosis other than AD (115/116). While we know that these diagnostic decisions were made, we have no information on whether they were ultimately appropriate, because there was no longitudinal follow-up to a postmortem gold standard diagnosis.
The diagnostic conclusions the physicians reached, based on their own unexplained judgment, would be consistent with very high negative and positive predictive values of the test. This could stem from a combination of factors: (1) the physicians’ acceptance of the high sensitivity and specificity for detecting amyloid in human brain, reported for the end-of-life population in Clark 2012; (2) an assumption that these performance characteristics apply to their current patients, who represent various (but undefined) subpopulations with cognitive impairment, but certainly not an end-of-life population as in Clark; and (3) that cognitive impairment plus a positive amyloid scan equals AD (e.g., a clear preference for one of at least two plausible hypothesis about the role of amyloid in AD development).
There is no empirical evidence internal to this study (or in prior studies) to support or explain the phenomenon of clinical decision making observed. This study appears to assume – but does not prove – such high negative and positive predictive values of the test (nor are these demonstrated in other studies, to our knowledge). This assumption may be implied by the authors themselves: “as AD is responsible for the large majority of cases of dementia with amyloid pathology (Barker 2002) physicians [in the study] may also be using their knowledge of the known clinical-pathologic correlations in making their diagnostic determinations” (Grundman 2012). These “correlations” have to do with the role of amyloid in AD; however, competing hypotheses of this role are vigorously debated in the literature.
An additional question about the decision making of the study physicians arises because their intended management does not always align with their revised, post-scan diagnosis. For instance, while 99% of subjects with a negative scan were given a final diagnosis of something other than AD, as pointed out by a MEDCAC panel member, approximately half of patients with a negative scan who were planned to get AD medications were still to receive them despite the negative scan (Grundman 2012, Table 5). Patients in the other half in this pool were no longer planned to get such medications as a result of the negative scan. The study did not explicitly discuss the reasons for these decisions, let alone quantitatively assess the likely harms supposedly avoided. As we discuss in more detail later, harm potentially exists if patients with FTD are mistakenly diagnosed with AD and placed on such medications.
The underlying design of the study produces an apparent circular logic: the scan is meaningful because its results alter diagnosis and management; but it does so appropriately only if one assumes its results are meaningful. This logic appears in other parts of the paper’s discussion section, for example:
“Changes in diagnosis occurred almost equally for subjects who had already undergone extensive evaluations (group A) and those in the middle of an ongoing diagnostic work-up (group B), arguing that in these patients, florbetapir PET scans provided potentially valuable information that seemed independent of other commonly performed diagnostic tests.”
In other words, because changes in diagnosis were (subjectively) made based on the scans, the scans must have provided valuable information.
As some MEDCAC panel members commented, the study “…raises more questions than it answers.” But this gets to its real value: it is a good hypothesis-generating study. It is possible that amyloid scans will someday meaningfully alter “the pattern of medication use, additional diagnostic testing, referral to AD resources, and clinical trial consideration.” We address the logic of, and evidence for, many of these possibilities when discussing “the value of a negative scan” later in this DM.
With respect specifically to the Grundman 2012 finding of decreased utilization of other tests, such as MRI and/or CT, we view this as a plausible hypothesis but one that has yet to be demonstrated. It is equally plausible that, even if PET Aβ imaging were widely available, most patients in the real world would continue to get MRIs and/or CTs anyway (to rule out other causes of, or contributors to, cognitive impairment, such as cerebrovascular disease, intracranial hemorrhage, and normal pressure hydrocephalus), ordered by other physicians, before the patient is evaluated by a dementia specialist. Perhaps more importantly, at least from a beneficiary’s perspective, given that radiation exposure in the elderly is less harmful than in younger populations, inappropriate imaging likely represents a much lower direct harm than being inappropriately placed on toxic medications.
Finally, there was no evaluation in Grundman 2012 of when amyloid imaging might be used instead of, or in combination with, other studies – or if it should be used at all – for particular patients. This foreshadows issues we will explore in detail later: what are the risk pools, how are they defined, what is the prevalence of disease in them, and what combination of tests are most appropriate for diagnosing patients in those pools? Answers to these questions are what are needed to define evidence-based coverage criteria for any given test – including PET Aβ imaging.
The meaning of a negative and positive scan
The Grundman 2012 study aside, there are other arguments and supporting evidence, presented by experts writing in the medical literature, speaking at the MEDCAC meeting, or in the NCD request itself, that are germane to the central questions of this NCD.
The core argument from many commenters is that although the gold standard for diagnosis of AD remains postmortem, and there is no cure or effective treatment for AD, there is value nonetheless to patient outcomes, directly or indirectly, in a negative scan. A negative study is “inconsistent with the diagnosis of AD,” as stated in the FDA-approved label, and this information could be useful to:
- effectively exclude AD in most patients, and therefore avoid potentially harmful and burdensome treatments for those who, if not for the scan, might be mistakenly diagnosed with AD;
- hasten clinical work up for a correct diagnosis that perhaps could be effectively treated; and
- improve the quality and efficiency of research to develop better treatments for AD, by selecting patients for clinical trials based on biological, rather than just clinical and epidemiological, factors.
Additionally, it is argued, there is a “value of knowing” that is not only intrinsic, but also directly linked to access to health care services and support which materially and substantially impact the patient’s quality of life. As discussed above, we consider both avoidance of harm and quality of life to be legitimate health outcomes, hence germane to national coverage decisions.
We examine the logic of, and evidence for, these arguments, as they connect to key sub-questions generated in part by MEDCAC panel discussions:
- What is the meaning of a negative and positive amyloid scan for a patient?
Does this depend on what risk pool, or subpopulation, a patient falls into? Have these been identified, and do they include Medicare beneficiaries?
- In what specific scenarios might the test meaningfully change patient management to improve health outcomes?
Would such outcomes likely be sustainable outside the expert clinical trial setting, in general community practice?
- Do evidence gaps exist, and if so, what clinical studies could be done to confidently close those gaps?
Assessing performance characteristics of the scan
Fundamentally, a physician orders a test in an attempt to identify the “true state” of the patient. Does the patient have the disease or not? If the true state is known, there is no clinical need for testing on the same question. Since the physician here is trying to determine whether or not the patient has AD, predictive values are more clinically relevant than sensitivity and specificity.
Both sensitivity and specificity are based on prior knowledge of the patient’s true state, diseased or non-diseased. Sensitivity asks what portion of diseased persons will be identified as positive. Specificity asks what portion of non-diseased persons will be identified as negative. Sensitivity and specificity are test characteristics that vary depending on the chosen cut-off between positive and negative. One can set the test cut-off point according to the desires of the user since there are inherent methodological tradeoffs between high sensitivity and high specificity, and thus one must consider the risks of having more false positive or false negative results. A receiver operating characteristic (ROC) curve is customarily used to illustrate this tradeoff.
Data on the sensitivity and specificity of PET Aβ imaging are prominent results in virtually all relevant clinical trials. Yet when clinical trials use different reference standards for determining these values, they mean different things and so the studies are not comparable. For instance, a study could compare (1) an F18 imaging agent to PIB in detecting amyloid plaque burden in living brain; (2) a given imaging agent to autopsy findings of amyloid burden; (3) results of an imaging agent to the clinical diagnosis of AD, or of MCI; or (4) results of an imaging agent to the gold-standard diagnosis of AD, which requires both (a) the presence of moderate to frequent Aβ plaques and neurofibrillary tangles on autopsy, and (b) clinical documentation of progressive dementia during life.
While the last comparison would be most informative, it has not to our knowledge ever actually been studied. The apparent purpose of studies undertaken for the FDA, which led to the publications of Clark 2011 and 2012, was never to diagnosis AD per se, but to assess the ability of florbetapir to identify amyloid plaque in human brain. Of interest, an initial plan to simply compare florbetapir to PIB PET Aβ imaging was rejected (by an FDA advisory committee) in favor of using autopsy findings as the appropriate reference standard – again not for diagnosis of AD, but for presence of amyloid in the brain.
Other studies that use clinical diagnosis as the reference standard are less useful as the reason amyloid imaging is being investigated in the first place is precisely because of known, systematic inaccuracies in the clinical diagnosis of AD.
On reviewing Clark 2012, along with the prior studies that led up to it, we do not doubt that amyloid imaging is safe in humans, and “efficacious” for detecting amyloid burden in the end-of-life population in which it was tested (consistent with FDA findings). However, the critique by Laforce and Rabinovici of PIB PET amyloid imaging is apposite to florbetapir PET imaging: “Technical and patient factors that could lead to false positives and false negatives are not clear. PIB binds to both diffuse and neuritic plaques (Lockhart 2007) (the latter being more common in normal aging), and the relative contribution of each to the in vivo signal has not been determined” (Laforce 2011).
Finally, there are a total of 59 subjects (with specificity determined by a subset of 20 subjects) imaged with a PET amyloid imaging agent that is both clinically-relevant and FDA-approved (florbetapir), who have autopsy correlation, representing an end-of-life population only. This is not enough to confidently determine sensitivity and specificity (and test and patient factors that could alter these) let alone, as discussed next, the positive and negative predictive values of the test, in different patient subpopulations.
Lack of positive and negative predictive values for the scan
In comparison to sensitivity and specificity, positive and negative predictive values (PPVs and NPVs) address a more clinically relevant question. In patients whose true states are unknown, what portion of those with a positive test actually have the disease? What portion of those with a negative test do not have disease? These predictive values depend on the prevalence of the disease in the tested population (with prevalence being the proportion of persons in a defined population at a given point in time who have the disease). If a test is applied to both a high risk and a low risk population, a positive result is more likely to be a true positive in the high risk population. Conversely, a negative result is more likely to be a true negative in a low risk population (Coulthard 2007). Further discussion and examples are available at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083733/.
When referencing the sensitivity and specificity from Clark 2012, the follow up study by Grundman 2012 (discussed at length above) said “florbetapir PET has been shown to be > 90% sensitive and specific for identifying subjects with moderate to frequent neuritic plaques, as assessed at autopsy within 1 year of scan.” Grundman does not quote the “100% specificity” reported by Clark.
Consider for the sake of illustration that – although this has never been demonstrated – the impact of the test in wider community practice (not just in the expert clinical trial setting) has an impressive 90% sensitivity and 90% specificity (using the postmortem gold standard as the reference). What does this mean for a particular patient who gets the test? As discussed above, this depends on the PPVs and NPVs of the test. But these values vary, depending on what defined risk pool the patient falls into, and what prevalence of AD exists in that pool.
Now consider that pool to be the general older American population, which has a prevalence of AD of approximately 12.5% (NIA 2013). The above 90% values for sensitivity and specificity would generate a > 98% NPV (the chance the patient does not have the disease if the test is negative) but a PPV of only about 56% (the chance the patient has the disease if the test is positive). In this case, a negative scan virtually excludes AD, echoing the FDA-approved label that “a negative scan is inconsistent with the diagnosis of AD.” But the meaning of a positive scan is unclear (also consistent with the FDA-approved label that a positive scan does not confirm the diagnosis of AD or other disease).
There has been extensive research for other diseases to define patient subpopulations at risk (risk pools) and their associated prevalence of disease (e.g., for thromboembolic disease, to evaluate the usefulness of diagnostic tests, such as a D-dimer). We note a similar path is emerging in AD-related research on the subtypes of MCI (discussed later). Other factors (e.g., age, genetic predisposition, comorbidities, cognitive reserve) complicate any subtyping schemata. As Laforce and Rabinovici argue: “not yet established is whether the threshold of [amyloid]-positivity should be adjusted based on demographic factors such as age (as is done when scoring plaques at autopsy) (Braak 1999) or genetic variables such as the ApoE4 genotype. Significantly, the relationship between amyloid and dementia is weaker in older versus younger individuals (Savva 2009). The positive predictive value of a positive amyloid scan in determining the cause of the dementia will therefore be lower in older individuals [e.g., the Medicare population]. In general, amyloid PET will be more useful in ruling out (given the high sensitivity to pathology) than in ruling in AD as the cause of dementia, since the detection of amyloid may be incidental or secondary to a primary, non-Aβ pathology in some cases . . .” (Laforce 2011).
Laforce’s last point brings up another issue: throughout our above discussion of statistical prediction we have been regarding performance characteristics of the test with respect to the presence of AD itself. However, these performance values apply only to the presence of amyloid in human brain, and that may not equate to AD per se. While there are competing views of what the presence of a given threshold of amyloid in human brain means, a leading hypothesis acknowledges that while amyloid plaques may be virtually necessary, they may not be sufficient, as either a trigger for or marker of the progressive dementia of AD.
The implications of a negative scan
The first part of that equation – that presence of amyloid plaques is virtually necessary – reflects the FDA-approved label that “a negative scan is inconsistent with the diagnosis of AD,” and is not the question that is before us in this NCD. (Note however that if the scan is performed too early, and is negative, this does not exclude subsequent amyloid plaque formation that later does reach a threshold for positivity – although this is unlikely to apply to those aged 65 and older, who comprise the vast majority (83%) of Medicare beneficiaries (http://www.statehealthfacts.org/comparetable.jsp?ind=294&cat=6&sort=431, accessed April 22, 2013)).
A question that is before us in this NCD is, given that a negative PET amyloid scan could virtually exclude AD in many patients, what is its clinical utility? We now turn to the arguments that “the value is in a negative scan.”
First, could a negative scan excluding AD avoid harm that would have otherwise occurred if patients were misdiagnosed with AD and given medications for symptoms that were in fact caused by other disease(s)? We have already discussed that we consider avoidance of harm to be an informative health outcome. Medications typically given to AD patients, such as memantine and cholinesterase inhibitors, are not AD medications per se. They do not prevent, cure or modify the disease process of AD or, for that matter, any known disease. They may offer moderate, temporary improvement to patients with cognitive and/or neuropsychiatric symptoms stemming from a variety of etiologies (TEC 2013). For instance, they have demonstrated efficacy in dementia with Lewy bodies (DLB) (Graff-Radford 2012); and are perhaps even more effective for DLB than for AD (Samuel 2000). Cholinesterase inhibitors may improve symptoms in Huntington’s disease and possibly vascular dementia (de Tommaso 2007, Kavirajan 2007, TEC 2013). In these particular cases, no additional harm appears to result from a misdiagnosis that places patients on such dementia medications.
It is primarily in differentiating frontotemporal dementia (FTD) and AD that potential for harm appears to exist (and this indeed was the example presented in the NCD request). Cholinesterase inhibitors have been shown to exacerbate symptoms in some patients with FTD, and use of memantine has correlated with greater functional and cognitive decline (TEC 2013, Kertesz 2008, Mendez 2007, Moretti 2004, Boxer 2012).
The differential of FTD and AD can be clinically challenging. Both are characterized by progressive dementia. AD typically begins with memory loss; FTD, with behavioral and language disturbances. AD is more likely in older persons; FTD, in younger. However, there is significant overlap such that patients with histopathology of FTD have often met the diagnostic criteria for AD during life (Varma 1999), and 10%-40% of patients diagnosed clinically with FTD are found to have AD by postmortem gold standard (Rabinovici 2011). Complicating the issue is that some individuals can have co-morbid disease.
CMS covered FDG PET in 2004 for use specifically in the differential of FTD and AD. The two diseases have relatively distinct patterns of hypometabolism on PET (predominantly temporoparietal in AD, and frontal and anterotemporal in FTD).
In a study of 45 subjects, Foster (2007) demonstrated that use of FDG PET in clinical assessment was more reliable and accurate in distinguishing FTD from AD than clinical assessment alone. Rabinovici 2011 was a head-to-head comparison of PIB amyloid versus FDG PET in the differential of AD and FTLD. Although there was a total of 110 subjects, only a small sample size (n = 22) had histopathology. For these 22 subjects, overall classification accuracy (using two visual and one quantitative techniques) was 97% for PIB (n = 12) and 87% for FDG (n = 10).
Second, could a negative amyloid scan improve the quality and efficiency of clinical trials to develop effective treatments for AD? The argument, articulated here by Laforce and Rabinovici but made by many, is that amyloid imaging could “improve clinical trial design by enrolling patients based on biological, rather than clinical, phenotype. This is a necessary first step for the development and testing of disease-specific therapies” (Laforce 2011). Laforce continues that “initial studies have found that requiring a positive molecular biomarker for inclusion will render AD clinical trials more efficient . . . .” Although some evidence suggests otherwise, most evidence, including similar use of diagnostics in trials for other diseases, and a recent European decision approving amyloid imaging for enrichment of clinical trials, suggests a promising role for amyloid imaging for this purpose (EMA 2011, Pearson 2012).
Third, could a negative scan also hasten the work up for other, potentially treatable diseases? Plausible arguments are made either way, but all lack conclusive evidence. An argument for answering “No” to this question is this. If you had a convincing clinical picture of AD, many experts agree the scan would not be needed (e.g., Johnson 2013). How physician concerns about liability would impact real-world decisions whether to get the test, if it were available, is an open question however.
Conversely, if you did not have such a convincing clinical picture, work up to exclude other, diagnosable and potentially treatable diseases should proceed anyway (as it would if an amyloid scan were negative). The unavailability of an amyloid scan does not change that logic.
An argument for answering “Yes” to this question derives from examples such as this (raised by a speaker at the MEDCAC): A patient with progressive cognitive impairment and a differential diagnosis of normal pressure hydrocephalus (NPH) versus AD was referred to a surgeon for a possible shunt, but the surgeon declined because the patient did not fit the typical criteria for NPH. The patient was thus given a presumptive diagnosis of AD. His cognitive impairment persisted for twelve years, after which he finally received an Amyvid scan, which was negative. See this example in its entirety from part 00109 -line 9 to part 00110- line 10 of the MEDCAC transcript found at http://www.cms.gov/Regulations-and-Guidance/Guidance/FACA/Downloads/id66d.pdf.
The evidence for such arguments, either way, is of limited persuasiveness, based almost entirely on clinical vignettes and case studies, which carry unmitigated risk of methodological bias and confounding, rather than on clinical trials.
The implications of a positive scan
Perhaps a greater challenge is that while a negative scan might be helpful or even just reassuring for many patients, if the scan happens to be positive for those very same patients, the meaning of this result is unclear, certainly much less clear than that of a negative scan.
McEvoy and Brewer (2012) present the following clinical scenario and analysis:
Given the high prevalence of AD and its devastating effects, there is a lot of anxiety among older individuals about developing this disorder, especially among those with relatives with the disease. Thus, minor slips in memory function, including those that are normal in healthy aging, can become an obsession, generating a vicious cycle in which a patient notices a slip in memory, becomes attuned to additional slips, and develops increasing anxiety about memory function, which itself may interfere with memory and memory testing. It is not uncommon to see cognitively unimpaired and, often, highly educated elderly patients presenting to the physician’s office debilitated by fear that they are developing dementia . . .
Imagine, then, adding to this patient’s clinical evaluation an assessment for amyloid pathology, with the hope that the patient will be one of the approximate 35-85% (dependent on age (Rowe 2010)) of cognitively healthy older individuals with a negative test. A negative test would relieve the patient’s fear of AD, since an absence of amyloid is inconsistent with a diagnosis of AD. However, this would not rule out other neurodegenerative disorders. A positive test would be even harder to interpret, since 20-65% (dependent on age) of cognitively healthy individuals can be expected to test positive for amyloid (Rowe 2010).
Given that elevated amyloid deposition is thought to precede development of cognitive impairment by more than a decade, we believe that findings of amyloid positivity in the absence of objective cognitive impairment would be irrelevant, and possibly harmful to the well-being of the patient. Even if future research were to demonstrate that all healthy older individuals with elevated amyloid eventually develop AD, an amyloid test cannot yet tell whether the patient will decline in the coming year or even in the coming decade; a positive test gives no indication of the phase of this slowly developing disease. For elderly patients especially, a warning sign loses all relevance if it can only suggest that cognitive impairment is likely to develop sometime in the next 10-20 years.
We agree with the authors’ reasoning, cited evidence, and concerns about real-world clinical impact. This concern is especially relevant given statements by some experts (including at the MEDCAC meeting) that they intend to use an amyloid scan in clinical practice to help make a positive diagnosis of AD (despite lack of empirical evidence of when and how to do this, and despite the inconsistency of such use with the FDA-approved label). However, we note that McEvoy and Brewer’s argument is explicitly about “findings of amyloid positivity in the absence of objective cognitive impairment.” Whether documentation of cognitive impairment opens a window for appropriate use is a topic we will return to later. McEvoy’s discussion is a good segue into the next issue, on the “value of knowing.”
The “value of knowing”
Expert speakers at the MEDCAC, public commentary, and numerous discussions in the literature have brought up the value to individuals and their families of definitively knowing they have AD. Patients were even described by clinicians as “being relieved” by knowing they had a diagnosis of AD. However, there are several limitations of this argument (including but not limited to the clinical meaning of a positive scan); we address these one by one.
First, the argument is clearly not generalizable. Given that there is no cure or effective treatment for AD, many do not “want to know.” In an international poll, the question (number 26) was asked: “In the future, a medical test might become available that would tell people before they had symptoms whether they will get Alzheimer’s disease in the future. If such a test became available, how likely do you think it is that you would get the test—very likely, somewhat likely, not too likely, or not at all likely?” In the U.S., only 29.5% responded “very likely,” while an additional 34.6% responded “somewhat likely.” Other, more recent polls have also made clear that answers to various related questions are variable.
We recognize that conclusions drawn from public opinion polls, even those done with statistically robust polling methodologies, are of questionable evidentiary value for fundamental questions about disease. At most we can conclude from them that patient and family responses to a diagnosis of AD are likely to vary, and we will need to rely on future empiric evidence to know this with greater certainty.
More importantly, implicit to the question is the assumption that the test is definitive. These poll responses cannot apply to PET amyloid scans as, again, the meaning of a positive scan is unclear. The complexity and uncertainty surrounding the science renders polling difficult. There are no polls, to our knowledge, where subjects were asked: “Would you want this scan if there is an X-Y% chance that you will be misdiagnosed with AD, based on the risk pool you fall into – which is itself unknowable as the criteria for such pools have yet to be clearly demonstrated – and by the way, here is the potential impact of being misdiagnosed with AD . . .”
Ultimately, we recognize that patients and families may make different decisions when a hypothetical scenario of disease becomes a real one. We can anticipate that these decisions will reflect the diversity of personal and cultural values in the population, including some, e.g., religious beliefs and prior family experience with illness, that are not readily studied in randomized clinical trials. Indeed we can envision the possibility that five different families will arrive at five different decisions, and that by some measure all five might be judged “appropriate” by various persons. Seeking answers to such questions will extend beyond the traditional boundaries of evidence based health insurance coverage. That does not diminish their ultimate importance to our beneficiaries, but it does alert us of the challenges of applying an evidence based review paradigm in this context.
Prognosis versus diagnosis
Doraiswamy 2012 connects to this “value of knowing” argument. As discussed previously, a key finding of this study was that, in the MCI population, 29% of those with positive scans, compared to 10% of those with negative scans, converted to clinically diagnosed AD. Some experts, including at the MEDCAC meeting, pointing to these data (and prior supporting studies), argue that patients with a positive scan and symptoms of MCI have AD, and it is just a matter of time before this manifests (Aisen MEDCAC presentation, Sperling 2011, Hardy 1991, 1992). So, along this line of thinking, why do roughly 33% of cognitively normal older individuals have significant amounts of amyloid in their brain? Because it is an indolent process. As with prostate cancer, many of these individuals will die with, rather than of, the disease.
A competing hypothesis is that “Aβ accumulation is necessary but not sufficient to produce the clinical manifestations of AD. It is likely that the cognitive decline would occur only in the setting of Aβ accumulation plus synaptic dysfunction and/or neurodegeneration” (Sperling 2011). Amyloid accumulation appears to plateau, and downstream neuronal lesions are required, and indeed better correlate with clinical severity of disease than does amyloid. In this competing view, while some of the infamous 33% with high Aβ and normal cognition may actually have AD but have just never manifested symptoms – and maybe never will in their lifetime – some, perhaps even the majority, may have simply not been “tipped” by other, distinct, downstream lesions that are necessary for AD, and perhaps never would be even if they lived longer. That is, they do not, and never will, have the disease.
In this light, the NIA-AA guideline authors conclude (and we agree) that “at this point, it remains unclear whether it is meaningful or feasible to make the distinction between Aβ as a risk factor for developing the clinical syndrome of AD versus Aβ accumulation as an early detectable stage of AD because current evidence suggests that both concepts are plausible” (Sperling 2011).
Some experts have even suggested that amyloid plaque formation could be the body’s protective mechanism to the (unknown) underlying disease process (Selko 2002, Lee 2004, Shankar 2008).
Returning to the Doraiswamy study, what this study demonstrates is the progression of symptoms to the clinical state of dementia, not the etiology(ies) driving that progression, because the endpoint is not autopsy, essential for the gold standard diagnosis of AD. Prognosis and diagnosis can be different things, and this study is really about the former.
So armed with this study, what do we really know? Not which individuals have AD. Thus an amyloid scan here would not inform the use of effective disease-specific treatments – again, if these existed. And if they did exist, and merely had mild adverse effects, such treatments would be tried on a host of symptomatic patients, and there might well emerge classifications of cognitive impairment and dementia based on whether individuals were susceptible or resistant to a given treatment. If so, and these treatments were efficacious for more than one etiology of cognitive impairment and/or dementia, the diagnosis of AD in itself would become less relevant.
Leaving diagnosis aside, and returning to the strong hand of the Doraiswamy study, prognosis, how might prognosis alone, as predicted by a positive amyloid scan, change one’s decision-making and management?
The study was not designed to test this (no one study can do everything), but even theoretically this is unclear – at least at 18 months, the limit of the study follow-up. The study reports a 29% chance of progression from MCI to dementia if the amyloid scan is positive, compared to a 10% chance of the same if it is negative. Say you are one of these patients who get a scan, your result is negative, and therefore you are in the 10% group. How would this change your (or your physician-advisor’s) decision to do or not do something? Put another way, if you knew you had a 29% chance of a very bad thing happening to you, and you could take some meaningful actions as a result for you and/or your family, would you now not take those actions because you had only a 10% chance of that fate? If there were a 29% chance the airplane you were about to board would crash, would you now board it because there was only a 10% chance? More longitudinal data could certainly alter these numbers, and provide clearer implications for rational decision making and management.
Mild cognitive impairment (MCI)
In reviewing the Grundman 2012 study we noted that it did not identify a potentially high-yield, objectively defined, target population. Fortunately, multiple other studies do: it is the MCI population. This was a key insight shared by MEDCAC panel members and expert presenters alike during the meeting. Deriving from research beginning in the 1990s, with the term coined in 1999, MCI lies between the cognitive changes of normal aging and dementia. Individuals with MCI experience memory loss (amnestic MCI) or loss of thinking skills other than memory loss (nonamnestic MCI), to a greater extent than expected for age, but without impairment of day-to-day functioning. Individuals with MCI are at increased risk for developing dementia (whether from AD or another etiology), but many do not progress to dementia, and some get better (Petersen 1999 and 2009, Wolk 2009, Hughes 2011, Ward 2012, Landau 2012, Sachdev 2012).
Both amnestic and nonamnestic MCI have subtypes of “single” and “multiple” domain. For example, a person without memory loss but with documented impairment in attention and concentration, and subtle impairment in visuospatial skills, would have multi-domain, non-amnestic MCI (Petersen 2009).
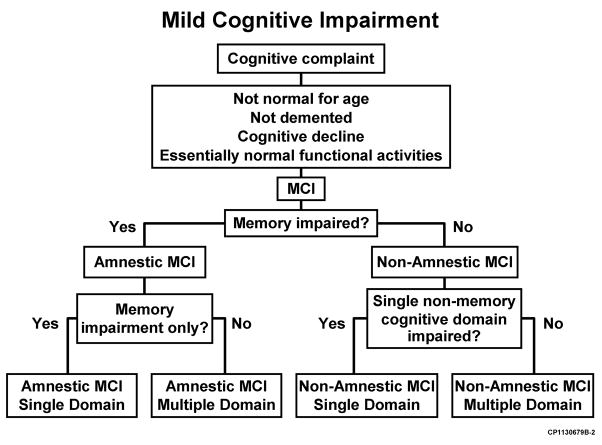
Figure obtained from Peterson, R. Early Diagnosis of Alzheimer’s Disease: Is MCI Too Late?
Current Alzheimer Research. 2009; 6(4):329.
More recent subtypes (under investigation in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Go and ADNI 2 trials) include “early” and “late” MCI. Early MCI represents subtle memory impairment that is intermediate between normal subjects and late MCI, as determined by say, education-adjusted scores on the Wechsler Memory Scale Logical Memory II (Landau 2012).
The field of MCI research is overlapping that of PET amyloid imaging, using various tracers including first PIB and increasingly florbetapir as well as other agents (Wolk 2009, Hughes 2011, Ward 2012, Landau 2012, Sachdev 2012, Cordell 2011). The ADNI family of prospective, longitudinal studies involve well over 1,000 participants at over 50 medical centers across the US and Canada, incorporate clinical classifications of MCI, AD and healthy controls, have regular, standardized clinical, imaging and CSF biomarker testing, and have autopsy as their endpoint. Other large, prospective, longitudinal studies of interest are underway at Mayo (Roberts 2008), in Australia (Sachdev 2012) and in Italy (Di Carlo 2007), although the degree of standardization that would enable meta-analysis across studies is not known to us at this time.
MCI subtypes, and associated objective scores on “bedside” mental status exams and neuropsychiatric testing, could, when combined with other patient characteristics (e.g., age, genetics, cognitive reserve, comorbidities) and biomarkers (for hypometabolism, plaque accumulation, synaptic dysfunction and neuronal loss), serve as the foundation for the development of objectively defined “risk pools,” or subpopulations of individuals who are at risk of progressing from MCI to AD. Ideally, risk stratification would eventually be able to identify persons at high risk for developing AD before symptoms occur. This may be especially important as a chain of evidence from multiple studies (animal and human) suggest that future therapies might be most (or only) effective if they begin early in or prior to the process of abnormal amyloid accumulation – perhaps10 to as much as 25 years prior to the onset of symptoms. Lifestyle changes, whether as a complimentary or an essential effort, may be a lifelong requirement (Gandy 2012, Goate 1991, Nicoll 2003, Bateman 2012, Jonsson 2012, Pollack 2012).
Generalizability
Generalizability – evidence that beneficial outcomes would be sustainable outside the clinical trial setting, in broad community use – is also a well-established factor that we consider in CMS coverage decisions. It is through this lens that we examine the questions of who should order, and who should interpret, PET Aβ imaging scans. We agree with the AIT that the ordering of PET Aβ imaging tests should be done by dementia specialists within the fields of neurology, neuropsychiatry and geriatric medicine who are actively managing the patient’s care (Johnson 2013).
As to the qualifications and training of physicians who would interpret (or “read”) the scans, we believe there is not enough evidence to support that the limited on-line training that currently exists suffices to ensure quality of reads in broader community practice. There are no experts we are aware of who do not acknowledge that this issue was a major problem with the initial launch of FDG PET, and we have learned from that experience as well as from the emergence of other new imaging technologies since then. A training and certificate model that may have some applicability for PET Aβ imaging is that for cardiac CT (Pelberg 2011). We believe that the training requirements included in the labeling should be viewed as absolute minimums and we encourage the development and maintenance of professional society standards. These might, for example, require formal mentoring of real cases and create facilitated pathways for “expert panel” interpretations of equivocal images.
Additionally, important questions remain about scan interpretation techniques themselves. Could quantitative measurements and visual interpretation be integrated by the reader (as done in say, CT brain perfusion imaging) to improve performance characteristics of the test? Should the anatomical distribution, as well as overall burden, of amyloid be considered in scan interpretation, especially given the discrepancies in frontal and medial temporal lobe findings between imaging and histopathology (Moghbel 2012, Kepe 2013). As mentioned earlier, PET amyloid tracers bind to both neuritic and diffuse plaques (Lockhart 2007), the latter being more common in normal aging, and the relative contribution of each to imaging results remains unclear. Also, it is unclear to what other substrates (Aβ structures, brain structures or receptors) these agents bind (Kepe 2013, EMA 2013 Annex 1). Finally, how could standardization – of PET generally (e.g., Wahl 2009) but also in amyloid imaging specifically – be improved to allow more meaningful comparisons across centers and trials?
In summary, we find that use of PET Aβ imaging is promising: (1) for excluding AD in narrowly defined and clinically difficult differentials, such as AD versus FTD, to prevent the harm of inappropriate use of potentially toxic medications; and (2) to improve the quality and efficiency of trials seeking to develop better interventions for AD, by allowing for selection of patients on the basis of biological as well as clinical and epidemiological factors. PET Aβ imaging may someday prove useful in limiting other testing, and, along with other biomarkers, in establishing a positive diagnosis of AD in certain subpopulations (to be defined), but the evidence to date is less substantial here. We also believe that further studies could be embedded into existing longitudinal, clinical research infrastructure, to potentially provide the building blocks for evidence-based appropriate use criteria. Finally, improvements in reading techniques, training and standardization of PET imaging protocols are needed.
B. CED
There are many outstanding questions about the diagnosis and management of AD and other dementias and the potential roles of PET Aβ imaging in that context. The goal of therapeutic trials may be to prevent, modify or cure the disease process, or to improve or slow the decline of patient cognition and functioning. Here the potential power of a negative scan to virtually exclude significant brain beta amyloid deposition could benefit Medicare beneficiaries, by helping them avoid potentially harmful, experimental therapies, and directing them to trials or treatments more likely to benefit them. Better patient selection could in turn improve the quality and efficiency of the therapeutic trials themselves. Due to the immense burden AD poses to Medicare beneficiaries (without considering burdens to their families and the Medicare system itself – which go beyond the scope of this NCD), the importance of developing effective therapies for AD rivals the difficulty of doing so.
While important questions on some ultimate outcomes may require comparison to autopsy findings that may not be available for years, other questions lend themselves to shorter time frames. For example, do community based physicians, relying on the result of scans interpreted by community based readers, consistently modify drug therapy to avoid certain adverse events? Are these adverse events actually avoided, or are the predictive values of imaging in these settings different or less reliable?
Some commenters suggest that the experience of CED for FDG PET for dementia and neurodegenerative diseases is relevant to the current consideration of CED for PET Aβ. They note specifically that a planned large trial of FDG PET has been minimally enrolled despite the passage of many years. While we believe there are lessons to be learned from that experience, we do not agree that the conclusion of those lessons is that CED should be abandoned in this important clinical area. We have, since that FDG PET NCD, formally articulated the CED paradigm in guidance convened the MEDCAC on CED. We have described our relationship with AHRQ, and AHRQ’s role in supporting CED.
As we noted above in the response to public comment, CED is not limited to a single trial that addresses every aspect of the CED question(s). We acknowledge that approvable CED protocols may address one or more aspects of the CED questions, and that nontraditional study designs, e.g. practical observational studies and registries, may be methodologically appropriate or even favored for some aspects.
Ongoing research initiatives such as the ADNI could provide much of the infrastructure for generating the evidence we seek. As stated at the outset of this discussion section, to date, no prospective, longitudinal data have emerged to provide sufficient evidence to conclude that the use of PET Aβ imaging would meaningfully improve health outcomes, directly or indirectly, for Medicare beneficiaries who have or are at risk for developing AD. However, it may be possible to embed within such infrastructure the studies needed to close evidence gaps identified in this DM, at the MEDCAC meeting, and in the literature. Indeed, some are underway. These would include prospective, controlled, longitudinal studies, with, where appropriate, randomization and autopsy as an endpoint. Hopefully, surrogate markers could be eventually identified to render unnecessary the longitudinal follow up to autopsy; what these surrogates might be remains unclear at this time however. These studies should focus not on what clinicians intend to do, but on actual management following objective protocols.
Risk pools might be objectively determined combining clinical MCI subtypes, for instance, with other clinical, imaging and laboratory biomarker testing (as described above). The prevalence of AD could then be determined for each risk pool (by gold standard), and this in turn, combined with more data points for estimating sensitivity and specificity, could generate quantitative negative and predictive values for biomarker tests, alone or in combination, for each pool. These predictive values would determine the meaning of a test result – and if the test should even be obtained in the first place – for a particular patient. Establishing the clinically utility of that test – its meaningful impact on patient management that can be linked to downstream processes that improve health outcomes – is also of course important.
It is possible that different combinations of biomarkers (again, of plaque accumulation, synaptic dysfunction, neuronal loss, hypometabolism, etc.) may be appropriate for patients in different pools. Further research could give weights to the partial and combined contributions of these various biomarker and clinical tests for specific risk pools. Identifying such pools, and the predictive values of diagnostic tests for each, has been essential for determining which individuals need what test, when, in clinical research of other diseases (such as thromboembolic disease, the example given earlier), where they have informed the development of evidence-based appropriate use criteria for diagnostic tests.
It is in this light that we assess the first iteration of the appropriate use criteria recently published by the joint Amyloid Imaging Taskforce (AIT) of the AA and SNMMI (Johnson 2013). It is a consensus statement. It does not delve into specifics about risk pools, their associated prevalence of disease, and the predictive values for various biomarker tests, alone or in combination, for each pool. It does not use these building blocks of evidenced-based appropriate use criteria, because these blocks themselves do not yet exist for amyloid imaging in AD.
With respect specifically to biomarkers, the AIT “did not consider other proposed diagnostic biomarkers for AD and therefore did not draw any conclusions as to the relative value of amyloid PET compared to CSF, MRI and FDG PET.” Yet the AIT acknowledges that “the appropriate use of amyloid PET requires knowledge of all relevant findings of clinical evaluations, laboratory tests and imaging relating how each component of the accumulated evidence should be weighed.” Our assessment of the current literature is that there is insufficient data to empirically determine the relative weights of those components. This conclusion echoes that of the authors of the NIA-AA guideline workgroups:
“There was a broad consensus within all three workgroups that much additional work is needed to validate the application of biomarkers for diagnostic purposes . . . additional biomarker comparison studies are needed, as is more thorough validation with postmortem studies, and the use of combinations of biomarkers in studies has been limited. Extensive work on biomarker standardization is needed before wide-spread adoption of these recommendations at any stage of the disease” (Jack 2011).
Knowing all this, the AIT’s approach seems to reflect the acceptance of certain premises: assuming the test will be used given FDA approval, and given the evidence that currently exists (as limited as it may be), what is the best guidance we can give to clinicians on how, and how not, to use this new technology? NCDs are inherently not guidance documents and thus reflect different premises. That said, we believe the AIT approach is informative and can help guide physician approaches to dementia management amid the challenges of an immature evidence base.
In its introduction, the AIT states that while “promising . . . experience with clinical amyloid PET imaging is limited. Most published studies to date have been designed to validate this technology and understand disease mechanisms rather than to evaluate applications in clinical practice. As a result, published data are available primarily from highly selected populations with prototypical findings rather than from patients with comorbidities, complex histories, and atypical features often seen in clinical practice. . . Empirical evidence for the value of added certainty resulting from amyloid PET has not yet been reported” (Johnson 2013).
This is consistent with CMS’ historic use of CED. We note in particular that the last sentence quoted above (with which we agree, based on our independent assessment of the literature) means it would be difficult for clinicians to be able to meet clause (iii) of the Preamble of the AIT’s appropriate use criteria:
“Amyloid imaging is appropriate in the situations listed here for individuals with all of the following characteristics: . . . (iii) when knowledge of the presence or absence of Aβ pathology is expected to increase diagnostic certainty and alter management” (Johnson 2013).
We believe emerging and future investigations, some of which are described in this DM, could no doubt better inform future iterations of the AIT’s guidelines.
We thus have finalized this decision as coverage with evidence development (CED). Many Medicare beneficiaries are potential candidates for AD-related therapeutic trials. Some therapies may prove successful in preventing or slowing the downstream cascade of neurodegeneration that correlates with severity of disease. However, we temper our enthusiasm as it also possible that future therapies, if they are effective at all, might be so only if used prior to or early in the process of amyloid accumulation. If the latter is the case, most patients who would benefit would be younger than age 65. We acknowledge that this would in turn create a healthier pool entering Medicare’s ranks; however, such dynamic, temporal analysis is outside the scope of our inquiry, which focuses solely on the Medicare population of today.
We have concluded that PET beta amyloid imaging is not reasonable and necessary under 1862(a)(1)(A) of the Act. However, CMS remains aware of significant evidence gaps that, if narrowed or closed, could further inform clinical decision making and future coverage policy. We believe Medicare could support this endeavor with CED. We have concluded that PET beta amyloid imaging is reasonable and necessary under 1862(a)(1)(E) of the Act.
Health Disparities
Subjects in key clinical trials on PET Aβ imaging (e.g., Clark 2011 and 2012, Grundman 2012) are generally > 90% white, despite data that older African-Americans are twice as likely, and older Hispanics 1.5 times as likely, to have AD (and other dementias) as older whites (see the Background section of this DM). This lack of evidence about racial and ethnic factors represents in our view an evidence gap that we encourage trial designers to consider when proposing clinical trial designs under this NCD. While recognizing that this consideration may complicate the design of appropriate clinical studies, we will nevertheless prefer clinical study proposals in which data on racial and ethnic factors are specifically collected and analyzed.
Summary
We have carefully and deliberately reviewed the available evidence, including published clinical studies, the MEDCAC recommendations, public comment and expert opinion, and we have reached the following answers to our analytic questions, which are repeated below for the convenience of the reader.
Question a:
Is the evidence adequate to conclude that PET Aβ imaging improves meaningful health outcomes in beneficiaries who display signs and symptoms of AD?
Answer a:
We cannot confidently conclude that PET Aβ imaging improves health outcomes in beneficiaries who display signs or symptoms of AD. We believe that additional clinical studies are needed to address these important issues, and that CED can facilitate this effort.
Question b:
Is the evidence adequate to conclude that PET Aβ imaging results inform the treating physician's management of the beneficiary to improve meaningful health outcomes? Those outcomes may include reasonably considered beneficial therapeutic management or the avoidance of unnecessary, burdensome interventions.
Answer b.
We have concluded from the available evidence that it is promising but not conclusive that PET Aβ imaging could, in community care settings, inform the identification of a specific population of beneficiaries in whom the harms of mismanagement with anticholinesterase therapy may be reduced if certain medications are in fact avoided.
CMS remains aware of significant evidence gaps that, if narrowed or closed, could further inform clinical decision making and future coverage policy. We believe Medicare could support this endeavor with CED.
IX. Conclusion
A. The Centers for Medicare & Medicaid Services (CMS) has determined that the evidence is insufficient to conclude that the use of positron emission tomography (PET) amyloid-beta (Aβ) imaging is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member for Medicare beneficiaries with dementia or neurodegenerative disease, and thus PET Aβ imaging is not covered under §1862(a)(1)(A) of the Social Security Act (“the Act”).
B. However, there is sufficient evidence that the use of PET Aβ imaging is promising in two scenarios: (1) to exclude Alzheimer’s disease (AD) in narrowly defined and clinically difficult differential diagnoses, such as AD versus frontotemporal dementia (FTD); and (2) to enrich clinical trials seeking better treatments or prevention strategies for AD, by allowing for selection of patients on the basis of biological as well as clinical and epidemiological factors.
Therefore, we will cover one PET Aβ scan per patient through coverage with evidence development (CED), under §1862(a)(1)(E) of the Act, in clinical studies that meet the criteria in each of the paragraphs below.
Clinical study objectives must be to (1) develop better treatments or prevention strategies for AD, or, as a strategy to identify subpopulations at risk for developing AD, or (2) resolve clinically difficult differential diagnoses (e.g., frontotemporal dementia (FTD) versus AD) where the use of PET Aβ imaging appears to improve health outcomes. These may include short term outcomes related to changes in management as well as longer term dementia outcomes.
Clinical studies must be approved by CMS, involve subjects from appropriate populations, and be comparative and longitudinal. Where appropriate, studies should be prospective, randomized, and use postmortem diagnosis as the endpoint. Radiopharmaceuticals used in the PET Aβ scans must be FDA approved. Approved studies must address one or more aspects of the following questions. For Medicare beneficiaries with cognitive impairment suspicious for AD, or who may be at risk for developing AD:
- Do the results of PET Aβ imaging lead to improved health outcomes? Meaningful health outcomes of interest include: avoidance of futile treatment or tests; improving, or slowing the decline of, quality of life; and survival.
- Are there specific subpopulations, patient characteristics or differential diagnoses that are predictive of improved health outcomes in patients whose management is guided by the PET Aβ imaging?
- Does using PET Aβ imaging in guiding patient management, to enrich clinical trials seeking better treatments or prevention strategies for AD, by selecting patients on the basis of biological as well as clinical and epidemiological factors, lead to improved health outcomes?
Any clinical study undertaken pursuant to this national coverage determination (NCD) must adhere to the timeframe designated in the approved clinical study protocol. Any approved clinical study must also adhere to the following standards of scientific integrity and relevance to the Medicare population.
- The principal purpose of the research study is to test whether a particular intervention potentially improves the participants’ health outcomes.
- The research study is well supported by available scientific and medical information or it is intended to clarify or establish the health outcomes of interventions already in common clinical use.
- The research study does not unjustifiably duplicate existing studies.
- The research study design is appropriate to answer the research question being asked in the study.
- The research study is sponsored by an organization or individual capable of executing the proposed study successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it must be in compliance with 21 CFR parts 50 and 56.
- All aspects of the research study are conducted according to appropriate standards of scientific integrity (see http://www.icmje.org).
- The research study has a written protocol that clearly addresses, or incorporates by reference, the standards listed here as Medicare requirements.
- The clinical research study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Trials of all medical technologies measuring therapeutic outcomes as one of the objectives meet this standard only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research study is registered on the ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject.
- The research study protocol specifies the method and timing of public release of all pre-specified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 24 months of the end of data collection. If a report is planned to be published in a peer reviewed journal, then that initial release may be an abstract that meets the requirements of the International Committee of Medical Journal Editors (http://www.icmje.org). However a full report of the outcomes must be made public no later than three (3) years after the end of data collection.
- The research study protocol must explicitly discuss subpopulations affected by the treatment under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations on the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The research study protocol explicitly discusses how the results are or are not expected to be generalizable to the Medicare population to infer whether Medicare patients may benefit from the intervention. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with §1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions.
All other uses are noncovered.
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service is reasonable and necessary. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes, to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to that group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is to the extent that differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a welldesigned and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of that have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
Randomized controlled trials
Non-randomized controlled trials
Prospective cohort studies
Retrospective case control studies
Cross-sectional studies
Surveillance studies (e. g., using registries or surveys)
Consecutive case series
Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in that confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to that the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and
route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in on-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude, and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.


