TO: Administrative File: CAG-00448N
FROM: Tamara Syrek Jensen, JD
Director, Coverage and Analysis Group
Joseph Chin, MD, MS
Deputy Director, Coverage and Analysis Group
Lori Ashby, MA
Director, Division of Medical and Surgical Services
Jyme Schafer, MD, MPH
Lead Medical Officer
Sarah Fulton, MHS
Lead Analyst
David Dolan, MBA, MA
Analyst
Rosemarie Hakim, PhD
Epidemiologist
SUBJECT: Proposed National Coverage Determination for Leadless Pacemakers
DATE: November 14, 2016
I. Proposed Decision
The Centers for Medicare & Medicaid Services (CMS) proposes to cover FDA-approved studies for leadless pacemakers through Coverage with Evidence Development (CED).
As a fully-described, written part of its protocol, the clinical research study must address the following research questions:
- What are the peri-procedural and post-procedural complications of leadless pacemakers?
- What are the long term outcomes of leadless pacemakers?
- What are the effects of patient characteristics (age, gender, comorbidities) on the use and health effects of leadless pacemakers?
CMS will review FDA-approved studies to determine if they meet the 13 criteria listed below. If CMS determines that they meet these criteria, the study will be posted on CMS’ CED website (https://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/index.html).
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries (RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint,even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
We are requesting public comments on this proposed determination pursuant to section 1862(l) of the Social Security Act. We are specifically interested in public comments on the use of CED in this decision. After considering the public comments, we will make a final determination and issue a final decision memorandum.
See Appendix B for our proposed manual language.
II. Background
Throughout this document we use numerous acronyms, some of which are not defined as they are presented in direct quotations. Please find below a list of these acronyms and corresponding full terminology:
ACC – American College of Cardiology
AF – Atrial Fibrillation
AV – Atrioventricular
AVB – Atrioventricular Block
CIED – Cardiac Implantable Electronic Device
CEC - Clinical Events Committee
CED – Coverage with Evidence Development
CMS – Centers for Medicare & Medicaid Services
CRT - Cardiac Resynchronization Therapy
DPIR – Danish Pacemaker and ICD Register
FDA – Food and Drug Administration
HRS – Heart Rhythm Society
ICD – Implantable Cardioverter Defibrillator
NCA – National Coverage Analysis
NCD – National Coverage Determination
NIS - Nationwide Inpatient Sample
PR – Pulse Rate
QOL – Quality of Life
RCT – Randomized Controlled Trial
SCAI – Society of Cardiovascular Angiography and Interventions
SND – Sinus Node Dysfunction
US – United States
VVI - Ventricular Pacing, Ventricular Sensing, Inhibition Response and Rate-adaptive
VVIR – Right Ventricular Pacing, Ventricular Sensing, Inhibition Response and Rate-adaptive
CMS initiated this national coverage analysis (NCA) to consider coverage under the Medicare program for leadless pacemakers.
The scope of this review is limited to leadless pacemakers. Traditional single and dual chamber pacemakers are not being reconsidered at this time and are therefore not within the scope of this NCA.
Symptomatic Bradycardia
Since the advent of the pacemaker in 1958, permanent cardiac pacing has become the standard treatment option for severe/symptomatic bradycardia, an arrhythmia indicating an unusually slow heart rate (typically less than 60 beats per minute), and is usually caused by sinus node dysfunction (SND) or atrioventricular blocks (AVB) (Mangrum and DiMarco, 2000; Udo et al., 2012). SND accounts for nearly 50% of pacemaker implantations in the United States, and can be attributed to intrinsic disease of the sinus node itself, or extrinsic causes such as medication (Mangrum and DiMarco, 2000). Bradycardia caused by SND occurs because there is abnormal automaticity in the sinus node, leading to failure of impulse formation or conduction from the node to the surrounding atrium.
AVB can also be attributed to intrinsic and extrinsic factors, and is divided into three degrees. First-degree AVB are frequently detected in electrocardiographs, and is defined as having a pulse rate (PR) interval over .2 seconds, while retaining a 1:1 AV relation (Mangrum and DiMarco, 2000). The PR interval is the “conduction time from the sinus node through the atrium, atrioventricular node, and His-Purkinje system to the onset of ventricular depolarization (Mangrum and DiMarco, 2000).” First-degree AV block does not cause bradycardia by itself, but can when seen observed with second or third-degree block or SND. Second-degree AVB can be Mobitz type 1 or Mobitz type 2, and “occurs when an organized atrial rhythm fails to conduct to the ventricle in a 1:1 ratio, but some atrial-ventricular relation is maintained (Mangrum and DiMarco, 2000).” Mobitz type 1 is observed when there is “a stable PP interval and a progressive increase in the PR interval until a P wave fails to conduct (Mangrum and DiMarco, 2000).” Mobitz type 2 second-degree AVB is diagnosed when “there is a stable PP interval with no measurable prolongation of the PR interval before an abrupt conduction failure (Mangrum and DiMarco, 2000).” Third-degree AVB refers to “complete heart block,” where atrial activity and ventricular activity occur independent of each other. Together, SND and AVB account for approximately 86% of pacemaker implantations in the United States (Bernstein and Parsonnet, 2001).
Treatment with Traditional Pacemakers
While there has been significant technological advancement in pacemaker technology over the past several years, pacemaker devices have continued to require the creation of a surgical pocket for the device to be implanted, and the use of leads to reach the pacing site (Ritter et al., 2015). The implantation procedure for traditional pacemakers is usually performed under local anesthesia and typically requires a brief hospitalization. The leads are threaded through a catheter that is inserted in the chest to the appropriate chamber(s) of the heart. The surgical pocket is made in the pad of flesh under the skin on the upper portion of the chest wall in order to hold the power source. This procedure is well validated, with 2.9 million pacemaker implantations performed between 1993 and 2009 in the United States alone (Greenspon et al., 2012).
Adverse Events following Traditional Pacemaker Implantation
Despite the several million pacemakers that have been implanted worldwide, serious adverse events remain a concern. In an analysis on data from the Dutch FOLLOWPACE study, which was designed to assess the frequency (and predictors) of short and long-term complications after first pacemaker implantation, Udo et al. (2012) found that 12.4% of patients developed complications at two months following implantation and nearly 16% of patients suffered complications at one year follow-up. This number rose to 18.3% at three years, and 19.7% five years after pacemaker implantation. The most frequent complications were lead-related, accounting for 5.54% of all complications within two months and long-term follow-up, with dislodgment of a lead being the most common adverse event (Udo et al., 2012). The second most frequent complications were pocket-related, such as hematoma and infection (4.75% of complications at two months, and 3.23% at long-term follow-up). Traumatic complications comprised the highest percentage of non-lead/pocket related complications (2.77% at two month follow-up), and consists of perforation of cardiac structure, pneumo(hemo)thorax, and pericardial effusion. Pulse generator problems accounted for the second most non-lead/pocket related complications (.33% within two months). During long-term follow-up, traumatic complications and pulse generator problems together accounted for 1.59% of adverse events. The full list of 2-month complications documented by Udo et al. (2012) can be seen in the table below:
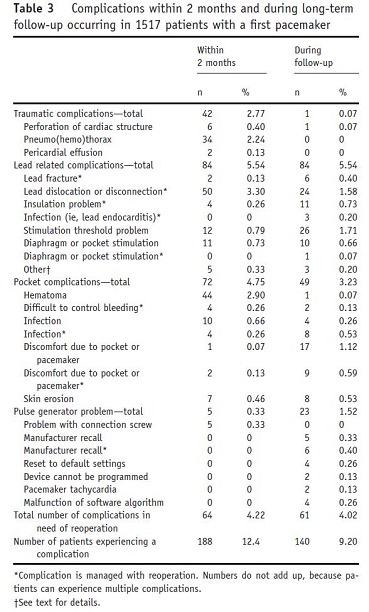
Page 731. Table 3. Complications within 2 months and during long-term follow-up occurring in 1517 patients with a first pacemaker. Udo et al. Incidence and predictors of short- and long-term complications in pacemaker therapy: the FOLLOWPACE study. Heart Rhythm. 2012;9:728-35.
Kirkfeldt, Johansen, Nohr, Jorgensen, and Nielsen (2014) examined complications stemming from cardiac implantable electronic device (CIED) procedures from May 2010, to April 2011 in all Danish patients. CIEDs included permanent pacemakers, cardiac resynchronization therapy (CRT) devices with defibrillators or without defibrillators, as well as implantable cardioverter defibrillators (ICDs). Patients were identified from the Danish Pacemaker and ICD Register (DPIR), and complications data was gathered from chart reviews. Complications were categorized as either major (all re-interventions (lead-related, infections, etc.), CIED-related systemic infections or endocarditis, pneumothorax requiring drainage, cardiac perforation, pocket revisions because of pain, deep venous thrombosis, Twiddler’s syndrome, wound revisions, stroke, myocardial infarctions, and procedure-related deaths) or minor (hematomas resulting in a prolonged hospital stay, hospital re-admissions/additional out-patient visits, wound infections treated with antibiotics, pneumothorax conservatively treated, and lead dislodgements that did not require re-intervention). Of the 5,918 patients that comprised the study population, 9.5% of these patients had experienced at least one complication, with 5.6% experiencing at least one major complication. Similar to the findings of Udo et al. (2012), the most common complication was the result of lead-related re-intervention, experienced by 2.4% of the patients studied (Kirkfeldt et al., 2014). The study also found one “possible” procedure-related death from a patient diagnosed with severe chronic obstructive pulmonary disease, who passed a few days after being discharged of an “unknown cause,” and had an unrecognized minor pneumothorax. No other patient deaths were indicative of procedure-related complications. While single and dual-chamber pacemakers accounted for 71% of the CIEDs in the study population, it is important to note the complications reported by Kirkfeldt et al. (2014) were not exclusive to pacemaker implantations.
Leadless Pacemaker Technology
Leadless pacemakers have the potential to significantly reduce serious adverse events related to pacemaker implantation by eliminating transvenous leads and the need for a surgical pocket. These devices are self-contained enclosed capsules that include the pacemaker electronics and battery, and currently range from around 26 mm to 42 mm in length, and have a maximum diameter between six mm and seven mm (Reddy et al., 2014; Ritter et al., 2015). Leadless pacemakers are delivered via catheter to the right ventricle of the heart, and function similarly to other transvenous single-chamber ventricular pacemakers. However, these devices have not been tested in broader, long term studies that include real world practice settings and are currently being followed in FDA-required post market studies.
III. History of Medicare Coverage
CMS does not have an existing NCD specific to leadless pacemakers. Coverage of leadless pacemakers is currently at the discretion of local Medicare Administrative Contractors (MACs) under §1861(a)(1)(A) of the Act.
Section 20.8 of the Medicare National Coverage Determinations (NCD) Manual is a long standing policy that dates back to 1983. In June 2000, Medtronic requested that CMS review the use of cardiac pacemakers to treat asymptomatic bradycardia in post-MI patients about to initiate long-term ß-blocker drug therapy. After a complete systematic review of the evidence provided, CMS posted a coverage decision memorandum (CAG-00063N) on March 20, 2001, that maintained the non-coverage NCD that pacemaker implantation would not be considered reasonable and necessary in these patients.
The NCD was updated in 2004 when CMS issued a decision memorandum to focus the NCD on the indications for pacemaker use rather than on the pacemaker implantation procedure. This reflected CMS’ finding that pacemaker implantation was no longer an experimental procedure. CMS only changed the framework of the NCD and did not review the specific provisions setting forth conditions that indicate that cardiac pacing is reasonable and necessary.
Effective August 13, 2013, CMS established conditions of coverage and non-coverage for permanent single chamber and dual chamber cardiac pacemakers in section 20.8.3 of the NCD Manual following a formal external joint request from the Heart Rhythm Society (HRS) and the American College of Cardiology (ACC) for reconsideration of dual-chamber pacemaker coverage.
The above NCDs did not consider evidence specific to leadless pacemakers; therefore coverage of leadless pacemakers does not fall under any of the existing pacemaker NCDs.
A. Current Request
CMS internally generated this NCA.
B. Benefit Category
For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act [§1812 (Scope of Part A); §1832 (Scope of Part B); §1861(s) (Definition of Medical and Other Health Services)].
Cardiac pacemakers qualify as:
- Inpatient hospital services.
- Physicians’ services.
- Prosthetic devices.
Thus, cardiac pacemakers qualify as a benefit.
Note: This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
IV. Timeline of Recent Activities
| Date |
Action |
| May 18, 2016 |
CMS internally generates and opens an NCA on leadless pacemakers. The 30-day public comment period begins. |
| June 17, 2016 |
Initial 30-day public comment period closes. |
V. Food and Drug Administration (FDA) Status
On April 6, 2016, the FDA approved the first leadless pacemaker device, the Medtronic Micra Transcatheter Pacing System (TPS). This device is indicated for use in patients who have experienced one or more of the following conditions:
- Symptomatic paroxysmal or permanent high-grade AV block in the presence of AF;
- Symptomatic paroxysmal or permanent high-grade AV block in the absence of AF, as an alternative to dual chamber pacing, when atrial lead placement is considered difficult, high risk, or not deemed necessary for effective therapy; and
- Symptomatic bradycardia-tachycardia syndrome or sinus node dysfunction (sinus bradycardia or sinus pauses), as an alternative to atrial or dual chamber pacing, when atrial lead placement is considered difficult, high risk, or not deemed necessary for effective therapy.
Rate-responsive pacing is indicated to provide increased heart rate appropriate to increasing levels of activity.
The complete FDA approval and labeling can be accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfTopic/pma/pma.cfm?num=P150033
VI. General Methodological Principles
When making national coverage determinations under section 1862(a)(1)(A) of the Social Security Act, CMS generally evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public comments sometimes cite published clinical evidence and give CMS useful information. Public comments that give information on unpublished evidence such as the results of individual practitioners or patients are less rigorous and therefore less useful for making a coverage determination. Public comments that contain personal health information will be redacted or will not be made available on the CMS website. CMS responds in detail to the public comments on a proposed national coverage determination when issuing the final national coverage determination.
VII. Evidence
A. Introduction
This presentation of evidence primarily focuses upon whether the pivotal randomized controlled trials (RCTs) are adequate to draw conclusions about health outcomes for leadless pacemakers, as well as whether the body of evidence is generalizable to the Medicare population. The evidence CMS examines has as its focus health outcomes, i.e., the benefits and harms of a particular treatment.
We summarize the evidence relating to the treatment of bradycardia with leadless pacemakers, which are currently being studied as right ventricular pacemakers (VVIR). The evidence includes two FDA premarket application clinical trials from two manufacturers. In treatment of these symptomatic arrhythmias, the primary focus is reduction in symptoms (chest pain, shortness of breath, fatigue and weakness), cardiovascular events (heart failure, stroke, myocardial infarction and arrhythmia) and mortality (cardiovascular mortality), as well as improvement in quality of life (QOL) and function.
Study endpoints should be clearly defined a priori to both improve the quality of clinical research and so as to allow comparison between clinical trials. Pacemakers are a substitute for the heart’s intrinsic pacing system and as such electrical variables and device function have been accepted for decades to support patient outcomes with traditional pacemakers. Leadless technologies present new challenges.
B. Discussion of Evidence
1. Evidence Question(s)
Is the evidence sufficient to conclude that treatment with leadless pacemakers improves health outcomes for Medicare beneficiaries with a slow heart rate (bradycardia)?
If the answer to the question above is positive, is the available evidence sufficient to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from leadless pacemakers?
2. External Technology Assessments
CMS did not commission an external TA for this NCA.
Kessels S, Parsons J, Ellery B from Adelaide Health Technology Assessment (AHTA). Health Policy Advisory Committee on Technology (HealthPACT). Technology Brief Leadless Pacemaker. July 2015.
“Summary of findings
Evidence for the safety and effectiveness of LPs is limited. Only one small study in humans has been finalised and the other trials assessing procedural and safety outcomes in patients indicated for single chamber ventricular pacing are still ongoing. It is not known if there are differences in effectiveness and safety when comparing the two devices (Nanostim and Micra). Data on the long term effectiveness and risk of complications, e.g. spontaneous dislodgement and embolisation, is lacking and should be investigated.
HealthPACT Advice
HealthPACT noted that this technology has the potential to reduce side effects and adverse events compared to pacemakers currently in use” and recommends “that the evidence be reviewed again in 24 months.”
Ndegwa S. Leadless pacemakers for the treatment of cardiac arrhythmias [Issues in emerging health technologies, Issue 134]. Ottawa: Canadian Agency for Drugs and Technologies in Health; 2015.
“Implementation Issues
…Although the setting and staff required for leadless pacemaker implantations do not differ from traditional pacemaker procedures, some training will be required for clinicians to become familiar with the device and implantation practices. The feasibility of device extraction in the case of battery depletion, device malfunction, or infection is currently unknown. Animal studies have shown that it is possible to retrieve the device six months after implantation, but it is not known if it is possible to safely retrieve the device after it has been in place for years. It may be possible to place multiple devices within the cardiac chamber, but it is not yet known if this is feasible.
Further evaluation of leadless pacemakers for long-term pacing performance” and complication rates “compared with traditional pacemakers is required. If long-term efficacy, safety, and cost-effectiveness can be demonstrated, leadless pacemakers may provide an additional treatment option for select patients with cardiac arrhythmias.”
3. Internal Technology Assessment
CMS searched PubMed for randomized controlled trials (RCTs), technology assessments, systematic reviews and clinical guidelines which featured or included leadless pacemakers; device complications; with keywords “leadless” and “pacemaker” Studies must have been published in peer-reviewed English language journals. Abstracts, voluntary registries, and studies with less than 50 patients (unless reporting a significant adverse event not published elsewhere) were excluded. The literature search was limited to the English language and specific to the human population, but included studies conducted in all countries, including the United States. Public access information from the FDA website was also used.
Reddy V., Exner D., Cantillon D., Doshi R., Bunch T., Tomassoni G., Friedman P., Estes N., Ip J., Niazi I., Plunkett K., Banker R., Porterfield J., Ip J., Dukkipati S., for the LEADLESS II Study Investigators. N Engl J Med 2015; 373:1125-35. doi: 10.1056/NEJMoa1507192.
This sponsor-funded study reports a planned interim analysis for this prospective single-arm multicenter U.S., Australian, and Canadian study. This interim analysis was performed on data from the first 300 patients who completed six months of follow-up in June 2015, with continuing enrollment. The primary efficacy end point was both an acceptable pacing threshold (< 2.0 V at 0.4 msec) and an acceptable sensing amplitude (R wave > 5.0 mV, or a value equal to or greater than the value at implantation) through six months. The primary safety end point was freedom from device-related serious adverse events through six months. Serious adverse events were defined as any untoward medical occurrence that led to death or to a serious deterioration in the health of a patient that resulted in life-threatening illness or injury, permanent impairment of a body structure or a body function, inpatient or prolonged hospitalization, or a medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function. An event was classified as device related in the clinical events-committee (CEC) attributed it to the procedure or device. Enrollment was continued after the first 300 patients, who were reported to have extended follow-up beyond six months. Additional outcomes were assessed in the 526 patients who were enrolled as of June 2015. Patients with indications for permanent single-chamber ventricular pacing, including chronic atrial fibrillation with atrioventricular or bifascicular bundle-branch block, sinus rhythm with second-degree or third-degree atrioventricular block and a low level of physical activity or a shortened expected life span, or sinus bradycardia with infrequent pauses or unexplained syncope with an abnormal electrophysioloical study. Additional inclusion criteria were: age > 18 years; life expectancy of at least 1 year; subject provides written informed consent; subject is not enrolled in another clinical study and is willing to comply with clinical investigation procedures and agrees to all required follow-up visits, test, and exams; subject is not pregnant and does not plan or is not planning to become pregnant during the course of the study. Patients were excluded if they had a mechanical tricuspid-valve prosthesis, pulmonary arterial hypertension, preexisting endocardial pacing or defibrillation leads, or an inferior vena cava filter or if they had undergone cardiovascular or peripheral vascular surgery within 30 days before enrollment.
The efficacy end point for device function was compared with performance goals based on data in an ongoing St. Jude pacemaker study (NCT01576016, “Accent Magnetic Resonance Imaging Pacemaker and Tendril Magnetic Resonance Imaging™ Lead Investigational Device Exemption Study”). As described in the study’s Supplementary Appendix, the performance goal for the safety endpoint was derived from two different pacemaker studies: the Ventricular Pacing, Ventricular Sensing, Inhibition Response and Rate-adaptive (VVI) pacemaker subset of 143 patients of a Finnish study (Pakarinen et al., 2010) (exact data for this subset was not published) and the FOLLOWPACE study a Dutch study of 1517 (Udo et al., 2012) patients at 2-month follow-up, removing any atrial lead complications. Enrolled patients had indications for single-chamber ventricular pacing, including chronic atrial fibrillation with AV or bifascicular bundle-branch block, sinus rhythm with second-degree or third-degree AVB and a low level of physical activity or a shortened expected life span, or sinus bradycardia with infrequent pauses or unexplained syncope with an abnormal electrophysiological study.
The device was successfully implanted in 504 of the 526 patients (95.8%). Some patients (29.8%) required repositioning after initial deployment, with 4.4% requiring repositioning more than two times. The intention-to-treat primary efficacy end point was met in 270 of the 300 patients in the primary cohort (90.0%; 95% confidence interval [CI], 86.0 to 93.2, P=0.007), with the lower boundary of the CI exceeding the prespecified calculated performance goal of 85%. The primary safety end point was met in 280 of the 300 patients (93.3%; 95% CI, 89.9 to 95.9; P<0.001), with the lower bound of the CI exceeding the performance goal of 86%. At six months, device-related serious adverse events were observed in 6.7% of patients. These events included device dislodgement with percutaneous retrieval in 1.7%, cardiac perforation in 1.3%, and pacing-threshold elevation requiring percutaneous retrieval and device replacement in 1.3%. Adverse events are listed in the table below.
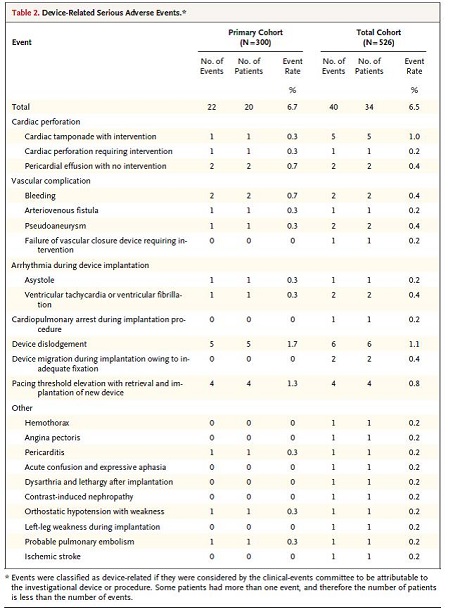
Table 2. Device-Related Serious Adverse Events. Page 1132. Reddy V., Exner D., Cantillon D., Doshi R., Bunch T., Tomassoni G., Friedman P., Estes N.., Ip J., Niazi I., Plunkett K., Banker R., Porterfield J., Ip J., Dukkipati S., for the LEADLESS II Study Investigators. N Engl J Med 2015; 373:1125-35. doi: 10.1056/NEJMoa1507192.
Deaths totaled 28 patients as listed in the table below. Two deaths were classified by the CEC as procedure related; none were deemed device related.
Causes of Death in the Total Cohort (N = 526); mean follow-up of 6.9 ± 4.2 months
| Cause |
No. |
| Cardiac |
|
| Arrhythmia |
2 |
| Heart failure |
1 |
| Unknown |
1 |
| Non-Cardiac |
|
| Accidental gunshot wound |
1 |
| Renal or liver failure |
5 |
| Respiratory failure |
3 |
| Multiple organ failure |
2 |
| Ischemic bowel/small bowel obstruction |
2 |
| Mixed respiratory and metabolic acidosis |
1 |
| Unknown |
10 |
| Total |
28 |
Source: Supplement to Reddy VY, Exner DV, Cantillon DJ, et al.
Percutaneous implantation of an entirely intracardiac leadless pacemaker.
N Engl J Med 2015;373:1125-35. doi: 10.1056/NEJMoa1507192.
http://www.nejm.org/doi/suppl/10.1056/NEJMoa1507192/suppl_file/nejmoa1507192_appendix.pdf
An analysis investigating the effect of operator experience on serious adverse events revealed a rate of 6.8% for the initial 10 cases versus 3.6% for subsequent implants, with an insignificant p value.
There were seven patients in the total cohort of 526 patients, excluding the six patients with dislodgements, where the pacemaker was successfully retrieved (range one to 413 days). The reasons for these retrievals were elevated pacing thresholds in four patients, worsening heart failure in two patients, and elective explant in one patient. The two patients with worsening heart failure received cardiovascular revascularization therapy (CRT) therapy.
A prespecified subgroup of 30 patients had 24-hour ambulatory electrocardiography (Holter monitoring). The mean minimum and maximum heart rates were 58.2+ 9.2 beats per minute and 111.1 + 21.1 beats per minute, with no mention of symptoms.
The authors state, "The leadless cardiac pacemaker met prespecified pacing and sensing requirements in the large majority of patients. Device-related serious adverse events occurred in approximately 1 in 15 patients."
Reynolds D., Duray G., Omar R., Soejima K., Neuzil P., Zhang S., Narasimhan C., Steinwender C., Brugada J., Lloyd M., Roberts P., Sagi V., Hummet J., Bongiorni M., Knops R., Ellis C., Gornick C., Bernabei M., Laager V., Stromberg K., Williams E., Hudnall J., Ritter P., for the Micra Transcatheter Pacing Study Group. A leadless intracardiac transcatheter pacing system. NEJM 2015; Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643.
This Medtronic-funded study reported a planned interim analysis for this prospective single-arm multicenter multi-nation study. Adjudications of adverse events were conducted by an independent clinical events committee. Enrolled patients met class I or II guideline-based indications for pacing (see Appendix C) (bradycardia due to atrial tachyarrhythmia, AV node dysfunction, or other causes), were considered to be suitable for single-chamber ventricular demand (VVI) pacing (ventricle paced, ventricle sensed, and pacemaker inhibited in response to a sensed best), able to undergo the study requirements, and were 18 years of age or older, if required by local law. Exclusion criteria included being entirely pacemaker dependent (defined as escape rhythm ≤ 30 bpm, though this restriction was removed after device reliability was verified), existing or prior pacemaker, ICD or CRT device implant, unstable angina pectoris, acute myocardial infarction within 30 days, current implantation or neurostimulator or any other chronically implanted device which uses electrical current, mechanical tricuspid valve, implanted vena cava filter, or left ventricular assist device, morbidly obese where telemetry communication of 12.5 cm cannot be obtained with programmer, femoral venous anatomy unable to accommodate a 23 French introducer sheath or implant on the right side of the heart, unable to tolerate urgent sternotomy, known intolerance to Nitinol, contraindication for single dose of 1.0 mg dexamethasone acetate, life expectancy < 12 months, enrollment in concurrent confounding study, and with pregnant or breastfeeding women. The primary safety end point was freedom from system-related or procedure related major complications. Major complications were defined as events resulting in death, permanent loss of device function as a result of mechanical or electrical dysfunction, hospitalization, prolongation of hospitalization by at least 48 hours, or system revision. The primary efficacy end point was the percentage of patients with low and stable pacing capture threshold at six months (< 2.0 V at a pulse width of 0.24 msec and an increase of < 1.5 V from the time of implantation).
These endpoints were evaluated against performance goals that were based on historical data from Medtronic dual pacemaker studies (with adjustments) and an analysis of Medtronic’s CareLink® database, respectively. A post hoc analysis was also performed in which the rates of major complications were compared in a control cohort of 2667 patients with transvenous pacemakers from six previously published studies of dual-chamber pacemakers. Detailed calculations of the performance goals and post-hoc historical comparison are available in the Supplementary Appendix in NEJM 2015; Feb 11;374(6). A total of 725 patients underwent an implantation attempt (720 was sample size for 90% power). The device was successfully implanted in 719 of 725 patients (99.2%). Primary patient indications were bradycardia associated with persistent or permanent atrial tachyarrhythmia (64%), SND (17.5%), AV block (14.8%), and other reasons. The reasons for the selection of ventricular pacing were indications associated with atrial tachyarrhythmia (65%), an expectation that pacing would not be frequent (29.7%), the patient’s advanced age (18.2%), and patient preference for new technology (12.3%). In 45 patients, leadless pacing was chosen because of conditions that precluded implantation of a transvenous pacemaker system. Patient characteristics:
_________________________
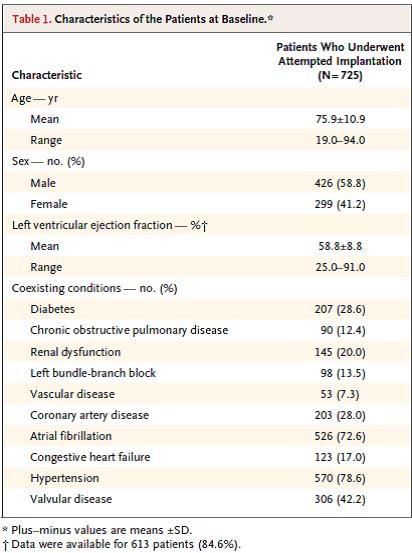
Table 1. Characteristics of the Patients at Baseline. Page 537. Reynolds D., Duray G., Omar R., Soejima K., Neuzil P., Zhang S., Narasimhan C., Steinwender C., Brugada J., Lloyd M., Roberts P., Sagi V., Hummet J., Bongiorni M., Knops R., Ellis C., Gornick C., Bernabei M., Laager V., Stromberg K., Williams E., Hudnall J., Ritter P., for the Micra Transcatheter Pacing Study Group. A leadless intracardiac transcatheter pacing system. NEJM 2015; Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643.
The Kaplan-Meier estimate of the rate of the primary safety end point at six months was 96.0% (95% CI, 93.9.97.3; p < 0.001 for the comparison with the safety performance goal of 83%); there were 28 major complications in 25 of 725 patients (all patients were analyzed for safety when the first 300 patients had reached six months of follow-up), and no dislodgements. There were 13 total cardiac injuries reported and adjudicated as related to the system or procedure and none resulted in death. “There was one death that was adjudicated as related to the transcatheter pacemaker implantation procedure. A 77 year old female patient had a concomitant procedure (AV nodal ablation) performed during the transcatheter pacemaker implantation, which resulted in prolonged procedure time. Of note, the patient had end stage renal disease and was scheduled for dialysis that day (it had been three days since the last dialysis session). No arterial blood gases were monitored during the procedure and no autopsy was conducted; however, the Investigator felt the most likely cause of death was metabolic acidosis due to prolonged procedure time with underlying end stage renal disease.” The major complications are listed below.
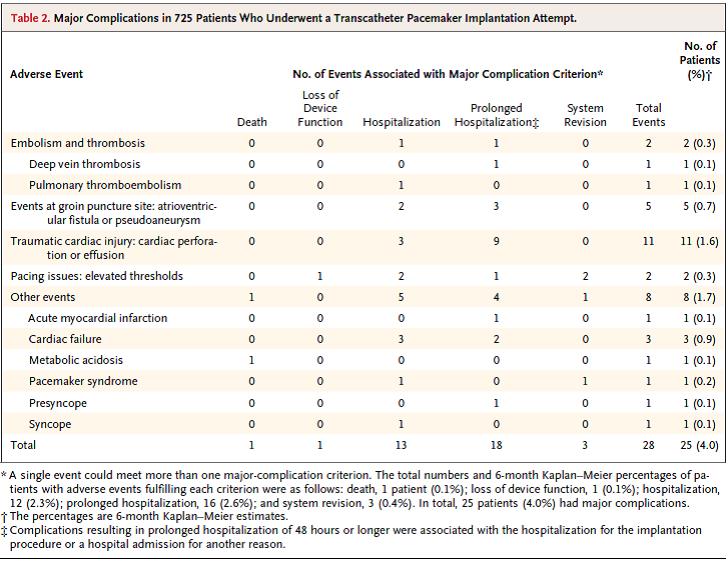
Table 2. Major Complications in 725 Patients Who Underwent a Transcatheter Pacemaker Implantation Attempt. Page 539. Reynolds D., Duray G., Omar R., Soejima K., Neuzil P., Zhang S., Narasimhan C., Steinwender C., Brugada J., Lloyd M., Roberts P., Sagi V., Hummet J., Bongiorni M., Knops R., Ellis C., Gornick C., Bernabei M., Laager V., Stromberg K., Williams E., Hudnall J., Ritter P., for the Micra Transcatheter Pacing Study Group. A leadless intracardiac transcatheter pacing system. NEJM 2015; Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643.
Causes of death in the total cohort
| Cause |
No. |
| Cardiac |
7 |
| Cardiac arrest |
1 |
| Cardiac failure |
5 |
| Pulseless electrical activity
| 1 |
| Non-cardiac |
22 |
| Abdominal injury |
1 |
| Respiratory failure/respiratory arrest |
2 |
| Bladder cancer |
1 |
| Chronic kidney disease |
2 |
| Chronic obstructive pulmonary disease |
1 |
| Dementia |
1 |
| Gastrointestinal hemorrhage/intestinal ischemia |
2 |
| Metabolic acidosis |
1 |
| Multi-organ failure |
2 |
| Pleural effusion |
1 |
| Pneumonia |
3 |
| Pulmonary embolism |
2 |
| Sepsis |
2 |
| Subdural hemorrhage |
1 |
| Total |
29 |
Source: Page 14. Table S5: Causes of death in the total cohort. Supplementary Appendix. Reynolds D., Duray G., Omar R., Soejima K., Neuzil P., Zhang S., Narasimhan C., Steinwender C., Brugada J., Lloyd M., Roberts P., Sagi V., Hummet J., Bongiorni M., Knops R., Ellis C., Gornick C., Bernabei M., Laager V., Stromberg K., Williams E., Hudnall J., Ritter P., for the Micra Transcatheter Pacing Study Group. A leadless intracardiac transcatheter pacing system. NEJM 2015; Feb 11;374(6):533-41. doi: 10.1056/NEJMoa1511643.
The rate of the primary efficacy end point (adequate 6-month pacing capture threshold) was 98.3% (95% CI, 96.1 to 99.5; p < 0.001 for the comparison with the efficacy performance goal of 80%) among 292 of 297 patients with paired 6-month data (only 301 had reached six month follow-up, six did not have paired data). The authors concluded, “In this historical comparison study, the transcatheter pacemaker met the prespecified safety and efficacy goals; it had a safety profile similar to that of a transvenous system while providing low and stable pacing thresholds.”
4. Medicare Evidence Development & Coverage Advisory Committee (MEDCAC)
Medicare did not hold a MEDCAC meeting on this topic.
5. Evidence-Based Guidelines
Epstein AE1, DiMarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Heart Rhythm Society. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation. 2013 Jan 22;127(3):e283-352. doi: 10.1161/CIR.0b013e318276ce9b. Epub 2012 Dec 19.
This document contains evidence based (wherever possible) recommended indications for pacing for bradycardia due to sinus and AV node dysfunction. The table below lists the major trials comparing atrial or dual-chamber pacing with ventricular pacing for the endpoints of quality of life or functional status, heart failure, atrial fibrillation, stroke or thromboembolism, and mortality.
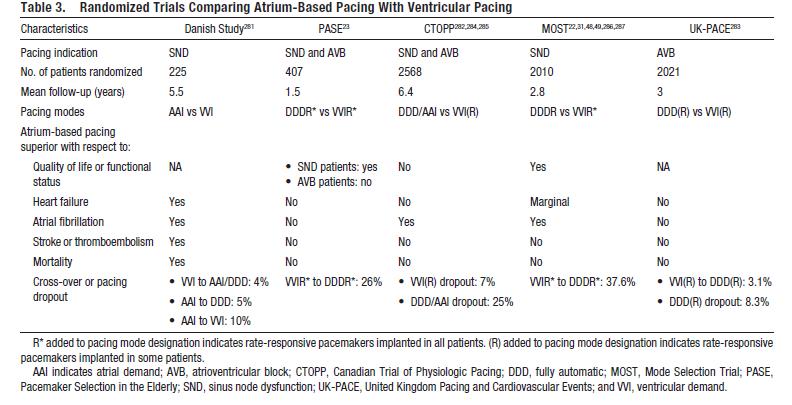
Table 3. Randomized Trials Comparing Atruim-Based Pacing with Ventricular Pacing. Epstein AE1, DiMarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Heart Rhythm Society. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society.Circulation. 2013 Jan 22;127(3):e283-352. doi: 10.1161/CIR.0b013e318276ce9b. Epub 2012 Dec 19.
6. Professional Society Recommendations / Consensus Statements / Other Expert Opinion
Gillis AM, Russo AM, Ellenbogen KA, Swerdlow CD, Olshansky B, Al-Khatib SM, Beshai JF, McComb JM, Nielsen JC, Philpott JM, Shen WK. HRS/ACCF expert consensus statement on pacemaker device and mode selection. J Am Coll Cardiol. 2012 Aug 14;60(7):682-703. doi: 10.1016/j.jacc.2012.06.011.
This document is a supplement to the guidelines document to assist in the selection of device type for patients who already meet guidelines for pacemaker implantation. Appendix C provides a table of these recommendations.
7. Public Comment
Initial Comment Period: 5/18/2016 – 6/17/2016
During the initial 30-day comment period, CMS received 46 comments, with three of these comments being omitted from publication on the CMS website due to excessive personal health information content, and one commenter posted twice. The majority of commenters were physicians/electrophysiologists, most of whom reported implanting leadless pacemakers themselves. One comment was submitted on behalf of joint professional societies which included the ACC, HRS, and SCAI. Four comments were made on behalf of groups, which included the Alliance for Patient Access, the Mayo Clinic Division of Cardiovascular Diseases, Partnership to Fight Chronic Disease, and the Society for Women’s Health Research. Three comments were from manufacturers of leadless pacemakers, while the three remaining comments were submitted by a nurse, a physician assistant, and the president of a consulting firm with experience regarding implantable medical devices.
All submitted comments supported coverage for leadless pacemakers, with the vast majority of comments referencing the elimination of complications stemming from the lead and pocket of traditional pacemaker implantations. Commenters also referenced the projection of longer battery life, reliable functionality, and quicker patient recovery times as reasons for supporting coverage. All initial public comments are available at https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=285.
VIII. CMS Analysis
National coverage determinations are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1869(f)(1)(B) of the Act). In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, with limited exceptions, the expenses incurred for items or services must be reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member." See §1862(a)(1)(A)of the Social Security Act.
In addition to §1862(a)(1)(A) of the Act, a second statutory provision may permit Medicare payment for items and services in some circumstances. That statute, section 1862(a)(1)(E) of the Act, provides, in pertinent part, that:
(a) Notwithstanding any other provision of this title, no payment may be made under part A or part B for any expenses incurred for items or services—
. . .
(1)(E) in the case of research conducted pursuant to section 1142, which is not reasonable and necessary to carry out the purposes of that section.
Section 1142 of the Act describes the authority of the Agency for Healthcare Research and Quality (AHRQ) to conduct and support research on outcomes, effectiveness, and appropriateness of services and procedures to identify the most effective and appropriate means to prevent, diagnose, treat, and manage diseases, disorders, and other health conditions. That section includes a requirement that the Secretary assure that AHRQ research priorities under Section 1142 appropriately reflect the needs and priorities of the Medicare program.
CED is a paradigm whereby Medicare covers items and services on the condition that they are furnished in the context of approved clinical studies or with the collection of additional clinical data. In making coverage decisions involving CED, CMS decides after a formal review of the medical literature to cover an item or service only in the context of an approved clinical study or when additional clinical data are collected to assess the appropriateness of an item or service for use with a particular beneficiary.
The 2014 CED Guidance Document is available at https://www.cms.gov/medicare-coverage-database/details/medicare-coverage-document-details.aspx?MCDId=27.
As noted earlier, our review sought answers to the questions below. We have repeated them here for the convenience of the reader.
Is the evidence sufficient to conclude that treatment with leadless pacemakers improves health outcomes for Medicare beneficiaries with a slow heart rate (bradycardia)?
If the answer to the question above is positive, is the available evidence sufficient to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from leadless pacemakers?
Permanent cardiac device-based pacing is the only effective treatment for symptomatic bradycardia. These devices reduce symptoms and syncope, and improve survival. Pacemakers for bradycardia can be of several different types: single-chamber right atrial-based; two-chamber right atria and right ventricle; right ventricular paced (VVIR). VVIR pacing has the most narrow guideline indication and is primarily for patients with permanent or persistent atrial arrhythmias and slow intrinsic heart rates.
Traditional permanent pacemaker technology has a subcutaneously implanted generator and transvenous leads and has been available for decades with few changes. The technology is well characterized through extensive bench testing experience and extensive market experience. Implant, fixation, and electrical data are well understood. The failure rate of these devices is predictable. Serious adverse events that occur with the current pacemakers are reported to be as high as 20% at five years, with significant contributions from the lead and pocket (Ritter et al., 2015; Udo et al., 2014). These complications include pneumothorax and hemothorax after subclavian vein puncture, pocket hematoma, erosion or infection, vein stenosis or occlusion, endocarditis, tricuspid valve trauma, lead fractures and other malfunctions (Ritter et al., 2015; Poole et al., 2010; Kirkfeldt et al., 2014; Greenspon et al., 2011; Johansen et al., 2011). Characteristics of independent predictors for complications have been studied, but the ability to predict who is at high risk is poor. An alternative that reduces complications is desirable.
Leadless pacemaker technology has the same inherent functionality as transvenous, single-chamber pacemakers. No leads or pockets are required. The technology is currently studied for right ventricle endocardium pacing. The devices are delivered to the right ventricle through the femoral vein and contain a reattachable mechanism for removal during the procedure. The devices are a self-contained capsule with a fixation mechanism and have rate-responsive functionality with an estimated longevity of 7 – 12 years.
The Micra® (Medtronic) device is currently FDA approved. The Micra® trial evaluated device efficacy and safety compared to performance goals based on historical data from recipients of conventional transvenous pacemakers. The functionality and features of this device are very similar to standard Medtronic pacemakers. The device met performance goals at pre-planned interim analysis of six months with 300 patients (efficacy data based on electrical parameters for 297 patients with paired data at six months, safety data for 725 patients though only 301 had reached the six month timepoint). The Micra® authors report that complications that led to death or that required invasive revision, termination of therapy, or hospitalization or extension of hospitalization occurred in 4.0% of the patients in the Micra® trial, in line with recent reports of transvenous systems (Kirkfeldt et al., 2014). There were 13 total cardiac injuries reported and adjudicated as related to the system or procedure and none resulted in death. No intervention was required in four, pericardiocentesis was conducted in seven, and surgical repair was required in two. While the traumatic cardiac injury was 1.6% in the study cohort as compared to 1.1% in a historical cohort, most of this accounted for in the difference in pericardial effusion (1.1% in the study group versus 0.5% in the historical control). Additionally, the study patients were older and had more coexisting conditions than did the control patients, and include a procedure learning curve. The study patients with cardiac injury were more likely to be elderly women with chronic lung disease or chronic obstructive pulmonary disease, however the authors stated the exact mechanism of these injuries was unclear. In a post hoc analysis, the 725 study patients were compared with the 2,667 patients who received transvenous pacemakers in the historical control cohort. Through six months of follow-up, the study patients had fewer major complications than did the patients in the historical control cohort. The patients in the study, as compared with patients in the control cohort, had significantly fewer hospitalizations and fewer system revisions due to complications. The rates of device or lead dislodgements were significantly higher in the control cohort than is the study cohort. Rates of access-site events, pacing issues, and cardiac injury events did not differ significantly between the cohorts. Battery longevity based on projections of conditions of use suggests a battery life of 12.5 years, similar to conventional pacing system batteries. Electrical performance characteristics of the device appear acceptable. Monitoring and diagnostic features are available with this device and it can be used in an MRI environment.
The LEADLESS II trial for the Nanostim™ (St. Jude) device (not FDA approved at the time of this publication) also met performance goals based on conventional transvenous pacemaker data, albeit compared to different historical data than the Micra® device. At six months, device-related serious adverse events were observed in 22 (6.7%) of the patients. The rates of cardiac perforation, device dislodgement, and elevated pacing thresholds necessitating device retrieval and replacement were 1.3%, 1.7% and 1.3%. Vascular complications were reported in 1.3% of the patients. In the total cohort of 526 patients, the rate of device related serious adverse events was 6.5% which included cardiac perforation in 1.5% of patients, device dislodgement in 1.1%, and device retrieval due to elevated pacing thresholds in 0.8%. Device migration to the pulmonary artery or right femoral vein occurred in four and two patients, respectively. While there were 28 deaths, four of those deaths were classified as having a cardiac cause. No deaths were considered to be device-related, however two deaths were classified by the CEC as procedure related. The first patient underwent successful implantation, but the procedure was complicated by a large groin hematoma followed by a significant drop in hemoglobin. Following stabilization, the patient was discharged from the hospital. At six day post-implant, a large area of ecchymosis was observed, the patient suffered a fatal cardiac arrest related to ventricular fibrillation fourteen days post-implant. This death was classified as cardiac and unrelated to the device, but was classified as related to the procedure. The second subject had tortuous venous anatomy and underwent an unsuccessful implant which was complicated by a pericardial effusion requiring pericardiocentesis. The subject went into atrial fibrillation with a rapid ventricular response two days after the attempted implant and was treated with antiarrhythmic therapy and cardioversion to restore sinus rhythm. The patient was discharged from the hospital in stable condition but presented again two days thereafter with onset of aphasia and found to be in atrial fibrillation with rapid ventricular rates and was found to have suffered a hemorrhagic stroke. The patient’s status continued to deteriorate and subsequently the patient’s status was changed to do not resuscitate. The patient died eight days after the failed implant and the death was judged to be procedure related.
There was determined to be an operator learning curve (6.8% device-related serious adverse events for the initial 10 cases versus 3.6% for the subsequent implants (p = 0.56)). Patient age and comorbidity demographics in both trials largely reflect the Medicare population though both studies were international. Differences in performance between the two devices are currently unknown.
In addition to the absence of leads and lead related complications, the device battery is a novel component of leadless pacemakers. However, in relation to intended real-world longevity, limited performance data is available. The fixation mechanism and implantation mechanism is new, with clinical performance reflected in the procedural data from 1251 trial participants. Device retrieval, replacement, and expiration present a different set of challenges, for which there is very little data. As a novel patient concern in comparison to current conventional pacemakers, remote monitoring is not currently available, so patients will be required to regularly visit their provider (Statement of American Heart Association to the Food and Drug Administration circulatory System Devices Panel, February 18, 2016).
Confidence in the current estimates of adverse events remains unclear and many questions remain unanswered. Problems with positioning/repositioning of the device have led to serious complications including death. The implant requires a different approach than that used for transvenous lead placement, with much larger venous access tools (sizes 18 and 23 French). Positioning the device in the apex of the heart has been associated with a deterioration in left ventricular function, though placement is not limited to the apex for either device (Tse and Lau, 1997; Lieberman et al., 2006). More information is needed regarding infection rates and remedies; the prevalence of device dislodgement (as a dislodged device will fail to function); the possibility for device extraction; long-term hemodynamic effects and thrombus formation issues; handling of end of battery life; safety of multiple leadless pacemakers; safety of shocks administered to patients with a leadless pacemaker; and variability in outcomes given periprocedural events observed (superior or inferior) based on operators and/or facilities. In summary, questions about health outcomes from broad leadless pacemaker use in the real world practice remain.
As with other VVIR pacemakers, the devices provide therapy for only a subgroup of patients who require pacemakers, perhaps fewer than 20% of all patients requiring a pacemaker. This type of pacing is generally used in patients with atrial fibrillation and bradycardia or in patients needing only infrequent pacing. Patients with many comorbidities who may not benefit from dual-chamber pacing and patients with no vascular access due to renal failure could be additional candidates. There is currently no role for patients with heart failure who are candidates for CRT. Potentially, many patients with a need for pacemakers will either have heart failure or will develop heart failure and will be CRT candidates. It remains unclear if providers will be able to confidently select appropriate patients given the paucity of data.
Remaining gaps in knowledge include real world safety and effectiveness, long term reliability and safety, battery longevity, device retrieval, device-device interactions, and best practices at device end of life (FDA panel presentation, 2016). There is no evidence-based shared decision making tool for this new technology, which raises a concern in how the risk and benefit profile of the procedure compared to traditional pacemakers will be presented to the patient.
Traditional pacemakers reduce symptoms and syncope, and improve survival, but these clinical outcomes are not available for leadless pacemakers. There is an indication of short term device function and short term procedural and device complications, but there is no data for chronic complications and long term performance. Due to the observational design without direct comparison to conventional pacemakers, the ability to draw conclusions about the relative performance and outcomes of these devices is limited. Six month outcomes are significantly different than the purported life of the device.
The available evidence is insufficient to confidently determine whether leadless pacemakers improve health outcomes in real world practice outside the controlled study environment. Evidence-based guidelines and professional society recommendations have also not specifically addressed leadless technologies to date. Thus CMS proposes that leadless pacemakers are not reasonable and necessary under §1862(a)(1)(A) of the Act.
Coverage with Evidence Development (CED)
Since the available evidence is promising, CMS proposes that leadless pacemakers should be covered through CED.
As a fully-described, written part of its protocol, the clinical research study must address the following research questions:
- What are the peri-procedural and post-procedural complications of leadless pacemakers?
- What are the long term outcomes of leadless pacemakers?
- What are the effects of patient characteristics (age, gender, comorbidities) on the use and health effects of leadless pacemakers?
Using the CED paradigm, we propose to cover FDA-approved post-market studies that have the following characteristics:
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries (RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint,even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
A major purpose of CED for leadless pacemakers is to fill remaining evidence gaps to help inform future coverage determinations. Evidence on the Medicare population and analyses specific to Medicare beneficiaries are important. A reconsideration of this NCD would require additional published evidence to inform the following remaining evidentiary gaps:
- What are the peri-procedural, and post-procedural complications, and long term outcomes of leadless pacemakers?
- Are leadless pacemakers equivalent or superior to conventional pacemakers in real world use?
- What are the infection rates, the long-term hemodynamic effects, and the rates of formation of thrombi?
- What are the patient demographics and effects of patient characteristics (age, gender, comorbidities) on the use and health effects of leadless pacemakers?
- What are the device-related issues (handling of end of battery life; effects of having multiple leadless pacemakers implanted; rate of device dislodgement; and the possibility of device extractions)?
- How are operators and facility characteristics related to peri-procedural and, post-procedural complications, and long term outcomes of leadless pacemakers?
Health Disparities
According to data from the Nationwide Inpatient Sample (NIS), from 1993 to 2009, the implantation of single-chamber ventricular (VVI) pacemakers dropped from 36% of all pacemaker implantations in the United States, to 14% (Greenspon et al., 2012). Over the same time period, the average age of patients receiving a VVI pacemaker rose from 77.5 years, to 80.1 years. In both premarket studies for the Nanostim™ and Micra® devices, the average age of patients receiving the leadless VVIR pacemaker was around 76 years. Considering the reported trend of a decreasing preference for VVI pacemakers coupled with the increasing age of recipients, examining the effectiveness of leadless pacemakers specifically in patients aged 80 and older would be useful.
While demographic information like patient age and comorbidity demographics in the Nanostim™ and Micra® device trials were largely representative of the Medicare population, it remains difficult to generalize the results to Medicare beneficiaries since both studies were international. Results from leadless pacemaker studies conducted in the United States are needed for Medicare beneficiaries to be properly represented. We recognize that leadless pacemakers are a new technology with limited studies to date. We will continue to monitor for disparities as additional evidence and patient use characteristics are developed.
Summary
Based on the above discussion, CMS believes that the evidence is promising and sufficient for coverage of FDA-approved leadless pacemakers for FDA-approved indications when furnished in accordance with FDA-approved protocols under CED. CMS believes that the available evidence is not sufficient to determine long term health outcomes or to identify the characteristics of the patient, practitioner or facility that predict which beneficiaries are more likely to experience overall benefit or harm from leadless pacemakers. Significant questions remain regarding the potential for deterioration in left ventricular function and other long term outcomes, as well as device longevity.
Coverage in the context of ongoing clinical research protocols or with additional data collection can expedite earlier beneficiary access to innovative technology while ensuring that systematic patient safeguards, including assurance that the technology is provided to clinically appropriate patients, are in place to reduce the risks inherent to new technologies, or to new application of older technologies. To ensure benefits to Medicare beneficiaries as leadless pacemaker technology grows and evolves, we are proposing coverage in FDA-approved studies under CED.
IX. Conclusion
The Centers for Medicare & Medicaid Services (CMS) proposes to cover FDA-approved studies for leadless pacemakers through Coverage with Evidence Development (CED).
As a fully-described, written part of its protocol, the clinical research study must address the following research questions:
- What are the peri-procedural and post-procedural complications of leadless pacemakers?
- What are the long term outcomes of leadless pacemakers?
- What are the effects of patient characteristics (age, gender, comorbidities) on the use and health effects of leadless pacemakers?
CMS will review FDA-approved studies to determine if they meet the 13 criteria listed below. If CMS determines that they meet these criteria, the study will be posted on CMS’ CED website (https://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/index.html)
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries(RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint,even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
We are requesting public comments on this proposed determination pursuant to section 1862(l) of the Social Security Act. We are specifically interested in public comments on the use of CED in this decision. After considering the public comments, we will make a final determination and issue a final decision memorandum.
See Appendix B for our proposed manual language.
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service is reasonable and necessary. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique
methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to that group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is to the
extent that differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well-designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of that have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
Randomized controlled trials
Non-randomized controlled trials
Prospective cohort studies
Retrospective case control studies
Cross-sectional studies
Surveillance studies (e. g. , using registries or surveys)
Consecutive case series
Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in that confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to that the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude, and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.
APPENDIX B
Medicare National Coverage Determinations Manual
Draft
This material represents a preliminary draft of Medicare's national coverage determination (NCD) for comment purposes only. The decision may be revised after the consideration of public comments and any additional evidence.
Table of Contents
(Rev.)A. General
The leadless pacemaker eliminates the need for a device pocket and insertion of a pacing lead which are integral elements of traditional pacing systems. The removal of these elements eliminate an important source of complications associated with traditional pacing systems while providing similar benefits. Leadless pacemakers are delivered via catheter to the right ventricle of the heart, and function similarly to other transvenous single-chamber ventricular pacemakers.
B. Nationally Covered Indications
Effective XX, XXXX, the Centers for Medicare & Medicaid Services (CMS) will cover FDA-approved studies for leadless pacemakers through Coverage with Evidence Development (CED).
As a fully-described, written part of its protocol, the clinical research study must address the following research questions:
- What are the peri-procedural and post-procedural complications of leadless pacemakers?
- What are the long term outcomes of leadless pacemakers?
- What are the effects of patient characteristics (age, gender, comorbidities) on the use and health effects of leadless pacemakers?
CMS will review FDA-approved studies to determine if they meet the 13 criteria listed below. If CMS determines that they meet these criteria, the study will be posted on CMS’ CED website (https://www.cms.gov/Medicare/Coverage/Coverage-with-Evidence-Development/index.html).
- The principal purpose of the study is to test whether the item or service meaningfully improves health outcomes of affected beneficiaries who are represented by the enrolled subjects.
- The rationale for the study is well supported by available scientific and medical evidence.
- The study results are not anticipated to unjustifiably duplicate existing knowledge.
- The study design is methodologically appropriate and the anticipated number of enrolled subjects is sufficient to answer the research question(s) being asked in the National Coverage Determination.
- The study is sponsored by an organization or individual capable of completing it successfully.
- The research study is in compliance with all applicable Federal regulations concerning the protection of human subjects found in the Code of Federal Regulations (CFR) at 45 CFR Part 46. If a study is regulated by the Food and Drug Administration (FDA), it is also in compliance with 21 CFR Parts 50 and 56. In addition, to further enhance the protection of human subjects in studies conducted under CED, the study must provide and obtain meaningful informed consent from patients regarding the risks associated with the study items and/or services, and the use and eventual disposition of the collected data.
- All aspects of the study are conducted according to appropriate standards of scientific integrity.
- The study has a written protocol that clearly demonstrates adherence to the standards listed here as Medicare requirements.
- The study is not designed to exclusively test toxicity or disease pathophysiology in healthy individuals. Such studies may meet this requirement only if the disease or condition being studied is life threatening as defined in 21 CFR §312.81(a) and the patient has no other viable treatment options.
- The clinical research studies and registries are registered on the www.ClinicalTrials.gov website by the principal sponsor/investigator prior to the enrollment of the first study subject. Registries are also registered in the Agency for Healthcare Quality (AHRQ) Registry of Patient Registries (RoPR).
- The research study protocol specifies the method and timing of public release of all prespecified outcomes to be measured including release of outcomes if outcomes are negative or study is terminated early. The results must be made public within 12 months of the study’s primary completion date, which is the date the final subject had final data collection for the primary endpoint,even if the trial does not achieve its primary aim. The results must include number started/completed, summary results for primary and secondary outcome measures, statistical analyses, and adverse events. Final results must be reported in a publicly accessibly manner; either in a peer-reviewed scientific journal (in print or on-line), in an on-line publicly accessible registry dedicated to the dissemination of clinical trial information such as ClinicalTrials.gov, or in journals willing to publish in abbreviated format (e.g., for studies with negative or incomplete results).
- The study protocol must explicitly discuss beneficiary subpopulations affected by the item or service under investigation, particularly traditionally underrepresented groups in clinical studies, how the inclusion and
exclusion criteria effect enrollment of these populations, and a plan for the retention and reporting of said populations in the trial. If the inclusion and exclusion criteria are expected to have a negative effect on the recruitment or retention of underrepresented populations, the protocol must discuss why these criteria are necessary.
- The study protocol explicitly discusses how the results are or are not expected to be generalizable to affected beneficiary subpopulations. Separate discussions in the protocol may be necessary for populations eligible for Medicare due to age, disability or Medicaid eligibility.
Consistent with section 1142 of the Act, the Agency for Healthcare Research and Quality (AHRQ) supports clinical research studies that CMS determines meet the above-listed standards and address the above-listed research questions. All other indications are nationally non-covered.
The principal investigator must submit the complete study protocol, identify the relevant CMS research question(s) that will be addressed and cite the location of the detailed analysis plan for those questions in the protocol, plus provide a statement addressing how the study satisfies each of the standards of scientific integrity (a. through m. listed above), as well as the investigator’s contact information, to the address below. The information will be reviewed, and approved studies will be identified on the CMS website.
Director, Coverage and Analysis Group
Centers for Medicare & Medicaid Services (CMS)
7500 Security Blvd., Mail Stop S3-02-01
Baltimore, MD 21244-1850
C. Nationally Non-Covered Indications
Leadless pacemakers are non-covered when furnished outside of a CMS approved CED study.
D. Other
NA
Appendix C: Consensus Recommendations
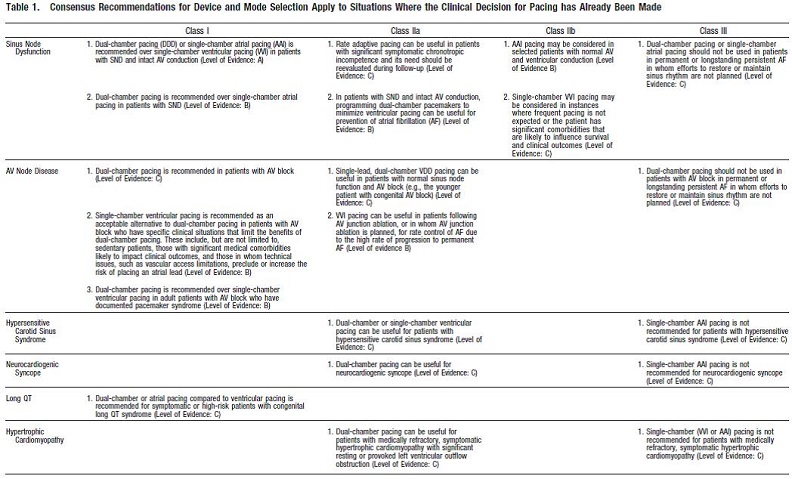
Table 1. Consensus Recommendations for Device and Mode Selection Apply to Situations Where the Clinical Decision for Pacing has Already Been Made. Gillis AM, Russo AM, Ellenbogen KA, Swerdlow CD, Olshansky B, Al-Khatib SM, Beshai JF, McComb JM, Nielsen JC, Philpott JM, Shen WK. HRS/ACCF expert consensus statement on pacemaker device and mode selection. J Am Coll Cardiol. 2012 Aug 14;60(7):682-703. doi: 10.1016/j.jacc.2012.06.011

Page 683. Classification of Recommendations and Level of Evidence. Gillis AM, Russo AM, Ellenbogen KA, Swerdlow CD, Olshansky B, Al-Khatib SM, Beshai JF, McComb JM, Nielsen JC, Philpott JM, Shen WK. HRS/ACCF expert consensus statement on pacemaker device and mode selection. J Am Coll Cardiol. 2012 Aug 14;60(7):682-703. doi: 10.1016/j.jacc.2012.06.011


