MRI for Patients with an Implanted Pacemaker, Implantable Cardioverter Defibrillator, Cardiac Resynchronization Therapy Pacemaker, or Cardiac Resynchronization Therapy Defibrillator
i) An MRI is covered when used according to the FDA labeling in an MRI environment for patients with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator.
ii) Any MRI for patients with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator that does not have FDA labeling specific to use in an MRI environment is only covered under the following conditions:
- MRI field strength is ≤ 1.5 Tesla;
- It has been ≥ 6 weeks since a patient’s device implantation or any lead revision or surgical modification;
- The patient is not pacemaker-dependent;
- The implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator system has no fractured, epicardial, or abandoned leads;
- The facility has implemented a checklist which includes the following:
- patient assessment is performed to identify the presence of an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator;
- before the scan, benefits and harms of the MRI scan are communicated with the patient or the patient’s delegated decision-maker;
- prior to the MRI scan, the implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator is interrogated and programmed into the appropriate MRI scanning mode;
- a qualified physician, nurse practitioner or physician assistant with expertise with implanted pacemakers, implantable cardioverter defibrillators, cardiac resynchronization therapy pacemakers, or cardiac resynchronization therapy defibrillators must directly supervise;
- a discharge plan that includes before being discharged from the hospital/facility, the patient is evaluated and the implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator reinterrogated to detect and correct any abnormalities that might have developed during the MRI.
See Appendix B for proposed manual language.
CMS recognizes that the tracking sheet informing the public that we are reconsidering coverage of MRI did not indicate our intention of only reconsidering the coverage with evidence development (CED) subsection (see 220.2(C)(1)) of 220.2 of the NCD Manual. The reason we were intending to only focus on section 220.2(C)(1) of the NCD manual was because there was a number of studies supporting MRI use for the indications in section 220(C)(1). Further, we recognize that there may be limitations to access as well as burden to patients and practitioners with the approved CED studies. We carefully reviewed all of the comments we received following the posting of the tracking sheet and note that several comments requested we expand to indications beyond our current NCD section 220.2(C)(1). We have addressed these comments in the analysis section of this NCA but did not propose any changes to any section of the NCD except 220.2(C)(1) of the NCD manual (with corresponding changes to add covered indications in 220.2(B)(3) for alignment). CMS is seeking comments on our proposed decision. We will respond to public comments in a final decision memorandum, as required by §1862(l)(3) of the Social Security Act (the Act).
II. Background
Throughout this document we use numerous acronyms, some of which are not defined as they are presented in direct quotations. Please find below a list of these acronyms and corresponding full terminology:
ACC – American College of Cardiology
ACR – American College of Radiology
AHA – American Heart Association
CED – Coverage with Evidence Development
CIED – Cardiovascular Implantable Electronic Device
CMS – Centers for Medicare & Medicaid Services
CRT-D - Cardiac resynchronization therapy defibrillator
CRT-P - Cardiac resynchronization therapy pacemaker
CT – Computerized Tomography
EMF – Electromagnetic Field
EMI – Electromagnetic Interference
FDA – Food and Drug Administration
HRS – Heart Rhythm Society
ICD – Implantable Cardioverter Defibrillator
IQR – Interquartile Range
MR – Magnetic Resonance
MRI – Magnetic Resonance Imaging
NCA – National Coverage Analysis
NCD – National Coverage Determination
PM – Pacemaker
US – United States
CMS initiated this national coverage determination (NCD) to reconsider coverage under the Medicare program for magnetic resonance imaging (MRI). MRI "is a noninvasive method of graphically representing the distribution of water and other hydrogen-rich molecules in the human body." Since its introduction into general practice in the 1970’s, MRI has been studied and used extensively in evaluation and management of many conditions such as cardiovascular and cerebrovascular diseases. Among its advantages are the absence of ionizing radiation, and the ability to achieve high levels of tissue contrast resolution, which allows characterization or diagnosis of lesions with or without use of contrast agents depending on lesion type. While a detailed discussion of MRI technology is beyond the scope of this decision, MRI is considered the gold standard for imaging patients with multiple sclerosis (MS) (Lohrke 2016, Polman 2011) to identify appropriate patients for beneficial treatments (Cochrane Reviews: La Mantia 2012, Tramacere 2015). MRI is also used in identifying candidates for coronary revascularization (Campbell 2014, Greenwood 2012, Jaarsma 2012) and is included in appropriate use of neuroimaging in the diagnostic workup of dementia (Health Quality Ontario 2014, Bermingham 2014). Concerns in patients with certain implanted cardiac devices have been reported and include pacing alterations, inappropriate ICD discharges, mechanical pull and rotation of the device have been reported (Schoenfeld, 2007).
III. History of Medicare Coverage
Section 220.2 of Chapter 1 of the Medicare National Coverage Determination (NCD) Manual, effective since 1985, established coverage of MRI for a number of uses. The policy has been expanded over the years; CMS last reconsidered this NCD in 2011 and established coverage with evidence development for patients with an implanted pacemaker or implantable cardioverter defibrillator.
Specifically, section 220.2(C)(1) currently describes contraindications:
C. Contraindications and Nationally Non-Covered Indications
- Contraindications
The MRI is not covered when the following patient-specific contraindications are present:
MRI is not covered for patients with cardiac pacemakers or with metallic clips on vascular aneurysms unless the Medicare beneficiary meets the provisions of the following exceptions:
Effective July 7, 2011, the contraindications will not apply to pacemakers when used according to the FDA-approved labeling in an MRI environment, or
Effective February 24, 2011, CMS believes that the evidence is promising although not yet convincing that MRI will improve patient health outcomes if certain safeguards are in place to ensure that the exposure of the device to an MRI environment adversely affects neither the interpretation of the MRI result nor the proper functioning of the implanted device itself. We believe that specific precautions (as listed below) could maximize benefits of MRI exposure for beneficiaries enrolled in clinical trials designed to assess the utility and safety of MRI exposure. Therefore, CMS determines that MRI will be covered by Medicare when provided in a clinical study under section 1862(a)(1)(E) (consistent with section 1142 of the Social Security Act (the Act)) through the Coverage with Study Participation (CSP) form of Coverage with Evidence Development (CED) if the study meets certain criteria (see NCD Manual).
A. Current Request
CMS opened this national coverage analysis (NCA) to reconsider coverage indications for MRI. We note that CMS’ intent regarding this MRI reconsideration was to only reconsider section 220.2(C)(1) rather than 220.2 of the NCD Manual in its entirety. We recognize that the tracking sheet did not indicate that CMS was only reconsidering CED (section 220.2(C)(1)). We have addressed any comments requesting additional modifications in sections other than 220.2(C)(1) in the public comment section of this NCA. After posting our tracking sheet, Russo and colleagues also submitted a request to reconsider section 220.2(C)(1) of the NCD.
B. Benefit Category
Medicare is a defined benefit program. For an item or service to be covered by the Medicare program, it must fall within one of the statutorily defined benefit categories outlined in the Social Security Act. MRI may be considered to be within the benefits described under sections: other diagnostic tests §1861(s)(3).
Medicare regulations at 42 CFR 410.32(a) state in part, that "…diagnostic tests must be ordered by the physician who is treating the beneficiary, that is, the physician who furnishes a consultation or treats a beneficiary for a specific medical problem and who uses the results in the management of the beneficiary’s specific medical problem."
This may not be an exhaustive list of all applicable Medicare benefit categories for this item or service.
IV. Timeline of Recent Activities
| Date |
Action |
| 07/12/2017 |
CMS opens an NCA for Initial 30-day public comment period begins. |
| 08/11/2017 |
First public comment period ends. CMS receives 17 comments. |
01/11/2018 |
Proposed Decision Memorandum posted. 30-day public comment period begins. |
V. Food and Drug Administration (FDA) Status
FDA granted approval of the first MR conditional pacemaker (Medtronic Revo MRI SureScanTM Pacing System) on February 8, 2011. Since that time, FDA approved MR conditional implantable cardioverter defibrillators (ICDs), cardiac resynchronization therapy defibrillators (CRT-Ds), and cardiac resynchronization therapy pacemakers (CRT-Ps) from various manufacturers. On April 6, 2016, FDA approved the first leadless pacemaker device (Medtronic Micra Transcatheter Pacing System (TPS)), which is also an MR conditional device.
FDA stated that these devices are MR conditional, meaning that certain criteria must be met for patients to get an MRI. For example, these conditions include performing scans in 1.5 Tesla (T) and in some cases 3.0T scanners. Similarly, the whole body specific absorption rate is typically limited to 2 W/kg (Normal Operating Mode). The conditions vary slightly across device manufacturers and are detailed in the FDA approved device labeling.
VI. General Methodological Principles
When making national coverage determinations, CMS generally evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service falling within a benefit category is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member. The critical appraisal of the evidence enables us to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for beneficiaries. An improved health outcome is one of several considerations in determining whether an item or service is reasonable and necessary.
A detailed account of the methodological principles of study design that the Agency utilizes to assess the relevant literature on a therapeutic or diagnostic item or service for specific conditions can be found in Appendix A.
Public comments sometimes cite published clinical evidence and give CMS useful information. Public comments that give information on unpublished evidence such as the results of individual practitioners or patients are less rigorous and therefore less useful for making a coverage determination. Public comments that contain personal health information will be redacted or will not be made available to the public on the CMS website. CMS responds in detail to the public comments on a proposed national coverage determination when issuing the final national coverage determination.
VII. Evidence
A. Introduction
CMS last reconsidered the MRI NCD (see Appendix C for § 220.2 of the NCD) in July 2011. CMS opened a national coverage analysis to reconsider the NCD based on more recent scientific evidence.
For this reconsideration, we reviewed the published medical literature from 2011 to 2017 to reassess the contraindications for those with a PM or ICD and to determine whether the coverage with evidence development (CED) questions have been answered. During our review, similar combination devices in cardiac resynchronization therapy pacemakers (CRT-Ps), or cardiac resynchronization therapy defibrillators (CRT-Ds) have been included in published studies and guidelines. These CRT-P and CRT-D devices are grouped together with PMs or ICDs (together also referred to as cardiovascular implanted electronic devices (CIEDs)) in published studies and guidelines, have identical considerations for MRI scans and have been included in our review, analysis and proposed decision.
Our evidence review only focused on whether to remove CED for implanted pacemakers (PMs), implantable cardioverter defibrillators (ICDs) and related combinations devices in cardiac resynchronization therapy pacemakers and defibrillators (CRT-Ps and CRT-Ds).
B. Discussion of Evidence
1. Evidence Questions
In assessing the evidence regarding the patient-specific contraindications for those with a PM or ICD (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators), our review and analysis of the evidence was guided by the following questions:
Q1. Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications informs the diagnosis or clinical management decisions in patients with implanted PMs or ICDs (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators)?
Q2. Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications improves health outcomes in patients with implanted PMs or ICDs (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators)?
The approved prospective clinical study must, with appropriate methodology, address one or more aspects of the following questions:
- Do results of MRI in PM/ICD beneficiaries with implanted cardiac devices affect physician decision making related to:
- Clinical management strategy (e.g., in oncology, toward palliative or curative care);
- Planning of treatment interventions; or
- Prevention of unneeded diagnostic studies or interventions, or preventable exposures?
- Do results of MRI in PM/ICD beneficiaries with implanted cardiac devices affect patient outcomes related to:
- Survival;
- Quality of life; or
- Adverse events during and after MR scanning?
2. External Technology Assessments
CMS did not request an external technology assessment (TA) on this issue.
3. Internal Technology Assessment
Literature Search Methods
CMS searched PubMed (MEDLINE and OVID) from July 2011 to October 2017. Search terms included: MRI, magnetic resonance imaging, ICD, defibrillator, and pacemaker. The search was limited to English language articles on studies with ≥ 20 participants, and excluded studies of MR conditional pacemakers, and those not involving human subjects.
We found 18 relevant studies including cohort studies, case-control studies, and case series analyses.
Awad, K., Griffin, J., Crawford, T. C., et al. (2015). Clinical safety of the Iforia implantable cardioverter-defibrillator system in patients subjected to thoracic spine and cardiac 1.5-T magnetic resonance imaging scanning conditions. Heart Rhythm, 12(10), 2155-2161. doi:10.1016/j.hrthm.2015.06.002
The aim of this study was to evaluate the clinical safety of the Biotronik Pro MRI Iforia ICD system during MRI.
This publication used data from a multi-center, prospective, single-arm, nonrandomized study which evaluated the clinical safety of the Biotronik ProMRI Iforia ICD system in 170 patients at 39 US centers undergoing 1.5 Tesla(T) MRI scanning of the thoracic spine (with no exclusion zone) or the heart. All devices were interrogated on enrollment, before and immediately after MRI, and one and three months post-MRI. The primary endpoints were (1) ventricular pacing threshold increase >0.5 V from pre-MRI to 1 month post-MRI; (2) R-wave amplitude decrease >50% from pre-MRI to one month post-MRI or R-wave amplitude < 5mV at one month post-MRI; and (3) MRI and ICD system-related serious adverse device effects.
The analysis method included exact binomial tests for primary endpoints utilizing an intention-to-treat (ITT) basis analysis, where the ITT population consists of all enrolled subjects who were programmed to the MRI mode before MRI scan.
Key exclusions were patients with a planned cardiac surgery within 3 months of enrollment; pregnancy; life expectancy ≥ 3 months; abandoned ICD or pacemaker leads; and implanted prostheses or medical devices that may complicate MRI studies.
The MRI protocol included: (1) interrogation of all devices upon study enrollment, before MRI; (2) during the scan patients were continuously monitored by ECG, pulse oximetry, and/or blood pressure monitoring; at least one physician, nurse practitioner, or physician assistant was present during the scan; (3) equipment and supplies needed to perform advanced cardiac life support were available (4) before the scan ICDs were placed into MRI mode, which disables VF detection/ therapy and pacing was programmed to either asynchronous mode or "off" at the physician’s discretion; (5) post-MRI, the ICD was interrogated, any pacemaker diagnostics recorded during scan were reviewed, the patient was assessed for adverse events, and programming was restored to initial parameters, including reactivation of ICD therapies.
Important patient demographics (sample size n=154) included mean age 60.0 years ±12.8 with a median of 59.8 years, 77.3% males, and ethnicity was unreported. A total of 153 patients underwent MRI (25.7% cardiac, 74.3% thoracic spine) and completed follow-up.
The study investigators found that freedom from the primary endpoints (i.e., did experience one of the abnormalities with regard to MRI) was met in all but one subject, in whom reduced R-wave amplitude was detected one month post-MRI. Reduced R-wave amplitude (4.7mV) was observed one month after programming to MRI mode although this subject did not undergo an MRI scan because of claustrophobia. No serious adverse device effects occurred during the course of the study. Ventricular pacing threshold did not increase >0.5 V in any participant (–0.01 ± 0.12V). Ventricular pacing impedance remained stable (–0.1 ± 38.9 Ω). P-wave amplitude and atrial pacing threshold were stable when pre-and one-month post-MRI values were compared (–0.075 ± 2.295 mV and 0.004 ± 0.140 V, respectively). Similarly, there was no significant change in atrial pacing impedance (5.0 ± 31.6 Ω).
The investigators concluded that this study provides evidence supporting the safety and efficacy of the Iforia ProMRI ICD system in patient undergoing cardiac or thoracic spine MRI without a scan exclusion zone. Further, they believe that regulatory changes are needed to allow MRI procedures to be performed with proper evaluation and monitoring by qualified personnel.
Cohen, J. D., Costa, H. S., Russo, R. J. (2012). Determining the risks of magnetic resonance imaging at 1.5 tesla for patients with pacemakers and implantable cardioverter defibrillators. Am J Cardiol, 110(11), 1631-1636. doi:10.1016/j.amjcard.2012.07.030
The aim of this study was to estimate the risk of MRI at 1.5 (T) for patients with cardiac devices (pacemakers and ICDs) by measuring the frequency of device failures and clinically relevant device parameter changes, as a pilot for a larger prospective registry.
This publication used data from a single-center, retrospective review of 109 patients with pacemakers and ICDs (the MRI group) who underwent 125 clinically indicated MRI studies at 1.5 (T) from February 2006 to March 2009 and compared them to data from a prospective cohort of 50 patients with cardiac devices who did not undergo MRI from August 2008 to June 2009 (the control group). The primary outcomes in the MRI group were: death, device or lead failure requiring immediate replacement, induced atrial or ventricular arrhythmias, loss of pacemaker capture, and electrical reset of the device (to default parameter settings), during the time interval of the MRI scan.
The analysis method included calculation of proportions and 95% confidence intervals for the primary outcomes. Linear mixed-model analyses were conducted to compare the MRI and control groups to compare the MRI and control groups with respect to battery voltage change, P- and R-wave percentage changes, high-voltage impedance change, pacing lead threshold, and impedance change, while adjusting for type of device (pacemaker or ICD), and pacemaker dependency (yes or no).
The MRI protocol included: (1) device interrogation performed immediately preceding the MRI study; (2a) in pacemaker dependent patients, the pacemaker was reprogrammed to an asynchronous pacing mode, and the magnet response was disabled when possible; (2b) in pacemaker dependent patients, pacing and sensing functions were deactivated; (2c) in ICD patients, tachyarrhythmia therapies were disabled; (3) patients were monitored throughout the procedure with continuous cardiac rhythm recording and pulse oximetry; (4) a cardiologist with experience in cardiac device programming who was able to place and use a temporary external cardiac pacemaker was present throughout the MRI study; (5) immediately after the MRI study, a repeat interrogation was performed using a protocol identical to the prescan interrogation, and prescan device parameters were restored.
The study included 22 cases of thoracic MRI (cardiac or thoracic spine). Important patient demographics (total sample size n=159 patients: 109 MRI group, 50 control group) included mean age 74 ± 11 years and 75 ± 10 years; 61% males and 64% males, respectively.
The study investigators found that no deaths, acute device failures, induced arrhythmias, losses of capture, or electrical reset episodes. Secondary outcomes focused on changes in device parameters that occurred during the MRI scan; of those differences between groups that were statistically significant, none led to clinically significant events.
The investigators concluded that MRI in patients with cardiac devices resulted in no device or lead failures. The authors acknowledged that this study "does not provide sufficient sample sizes or adequate follow-up to recommend changes to the current clinical guidelines for conducting MRI of patients with cardiac devices. However, the purpose of this retrospective study was to gather information to guide a larger prospective registry of patients with devices who would undergo clinically indicated MRI."
Friedman, H. L., Acker, N., Dalzell, C., et al. (2013). Magnetic resonance imaging in patients with recently implanted pacemakers. Pacing Clin Electrophysiol, 36(9), 1090-1095. doi:10.1111/pace.12213
The aim of this study was to evaluate the safety of MRI in patients with recently implanted pacemakers.
This study used data that was prospectively collected after implementation of an MRI safety protocol for patients with cardiac devices at the Mayo Clinic Heart Rhythm services and radiology service in January 2008. Retrospective analysis compared data from eight scans in patients with recently implanted (≤42 days) to 211 in non-recently implanted (>42 days) pacemaker leads.
Key exclusions were patients with <18 years of age; pacemaker dependence; presence of more than one implanted pulse generator; evidence of inadequate pacemaker function; abnormal baseline Troponin-I (TNI > 0.03 ng/mL) and/or creatine kinase-MB (CK-MB >6.2); requires continuous intravenous medication, especially for cardiac support.
The MRI protocol included: (1) the patient’s intrinsic rate being determined before scanning, the pacemaker was programmed to asynchronous pacing, and if the intrinsic rate was above 90 beats/min, the device was programmed to a monitor-only mode; (2) during the MRI, a radiologist, an MRI physicist, and a heart rhythm cardiologist or nurse specialist were present; (3) patients were monitored by a cardiologist or a pacemaker nurse throughout the MRI examination using pulse oximetry, CO2 measurement, and electrocardiography; (4) patients were asked if they felt any pain or discomfort following the MR scan; (5) post-MRI the device was re-interrogated for the same measurements as mentioned previously.
The analysis method included use of Generalized Estimating Equation models to try to account for the potential correlation from MRIs from the same patients to compare early versus late implants and pre-MRI versus post-MRI parameters.
Important patient demographics (sample size n=171) included a 58% male study population; no other demographics were reported in the publication.
The study investigators found that there were no clinically significant events and no statistically significant change in device parameter measurements between the two groups. "In one patient imaged 79 days post-implant, frequent premature ventricular complexes were noted during the scan, requiring no action."
The investigators concluded that "with a strong clinical indication and with careful monitoring, MRI imaging is feasible in patients with recently implanted pacemakers, although experience is limited."
Gold, M. R., Sommer, T., Schwitter, J., et al. (2015). Full-Body MRI in Patients With an Implantable Cardioverter-Defibrillator: Primary Results of a Randomized Study. J Am Coll Cardiol, 65(24), 2581-2588. doi:10.1016/j.jacc.2015.04.047
The aim of this study was to evaluate the safety and efficacy of a novel ICD system specially designed for full-body MRI without restrictions on heart rate or pacing dependency.
This study used data from a multicenter, 2:1 randomized trial evaluating the safety and efficacy of the Evera MRI ICD (MR-ICD, Medtronic) connected to commercially-available defibrillator leads (model 6935M or 6947M [Medtronic], 55- and 62-cm lead lengths) specially designed for full-body MRI without restrictions on heart rate or pacing dependency. The study was conducted from April 17, 2014,to September 11, 2014 across 42 centers located in 13 countries within North and South America, Europe, Asia, and the Middle East. Subjects received either a single- or dual-chamber ICD. The primary safety endpoint was freedom from a composite of MRI-related events within 30 days post-MRI. The primary efficacy endpoints were ventricular pacing capture threshold and ventricular sensing amplitude.
The analysis method included use of a one-proportion binomial exact test for the primary safety objective. The Farrington-Manning test of 2 independent proportions was used to test the primary efficacy endpoints. Mean change was tested using paired Student t-tests. Continuous variables are reported as mean ± SD.
The MRI protocol included: (1) pulse oximetry, electrocardiography, and verbal communication monitoring during the MRI; (2) an external defibrillator being immediately available during the MRI; (3) having personnel present to manage any potential emergency situation.
Important patient demographics (sample size n=263: 175 MRI group, 88 Control Group) included mean/median age 60.4 ± 13.8 years, 76% male, ethnicity was not available.
A total of 263 patients across 42 centers were randomized 2:1 to MRI of the chest, cervical, and head regions at 1.5T (n = 175), or to a one-hour waiting period without MRI (control group, n = 88).
The study investigators found that the safety endpoint was met with 100% freedom from the composite endpoint. Both efficacy endpoints were met with minimal differences in the proportion of MRI and control patients who demonstrated a <0.5 V increase in ventricular pacing capture threshold (100% MRI vs. 98.8% control) or a ≤50% decrease in R-wave amplitude (99.3% MRI vs. 98.8% control). The average MRI group VPCT did not change (0.00 ± 0.16 V) and the control group has a small average change (0.02 ± 0.16 V). Mean changes were small for both MRI (–0.10 ± 2.67 mV) and control (0.04 ± 2.59 mV) groups. A total of 34 ventricular tachyarrhythmia/fibrillation episodes (20 induced; 14 spontaneous) occurred in 24 participants’ post-MRI, with no observed effect on sensing, detection, or treatment.
The investigators concluded that the data provide evidence supporting that, "the system is safe with MRI examinations, showing no evidence of any adverse effect on the electrical performance or the ability to treat ventricular arrhythmias".
Higgins, J. V., Sheldon, S. H., Watson, R. E., et al. (2015). "Power-on resets" in cardiac implantable electronic devices during magnetic resonance imaging. Heart Rhythm, 12(3), 540-544. doi:10.1016/j.hrthm.2014.10.039
The aim of this study was to evaluate the frequency of, and risk factors for, power-on reset (PoR) in patients with non-MRI-conditional pacemakers undergoing MRI. Electromagnetic interference (EMI) during MRI could cause PoR and reversion of the CIED to its factory default settings. This could lead to inappropriate inhibition of pacing in patients with pacemakers, resulting in asystole, or inappropriate antitachycardia pacing or shocks in patients with ICDs.
This study used data prospectively collected at Mayo Clinic, Rochester, MN, between January 2008 and May 2013 in patients with non–MRI-conditional CIEDs undergoing clinically indicated MR.
The analysis method included use of descriptive statistics (mean ±SD) for normally distributed continuous variables, as median and interquartile range (IQR) for non-Gaussian distributed continuous variables, or as number and percentage for categorical variables. Between group comparisons were made using the Pearson χ2 test for categorical variables and 2-sample t test or Wilcoxon rank-sum test for continuous variables.
Key exclusions were patients who were pacemaker dependent, <18 years old, who had abnormal cardiac biomarkers, who required general anesthesia for MRI, or who needed a continuous intravenous infusion of a medication during MRI.
The MRI protocol included: (1) devices being programmed to an asynchronous mode or an inhibition mode, with tachyarrhythmia therapies off in patients with implantable cardioverter-defibrillators; (2) monitoring by an Advanced Cardiovascular Life Support–certified cardiac device nurse during the MRI; (3) continuous pulse oximetry, electrocardiography, and blood pressure; (4) staff radiologist and a radiology MRI physicist being present for the MRI; (5) visual and voice contact was maintained with the patient at all times during MRI to identify if the patient is experiencing pain, discomfort, or other perceived abnormality during the MRI; (6) devices were interrogated after all examinations and reprogrammed to their pre-MRI settings after the study.
A total of 256 MRI scans were performed in 198 patients with non-MRI-conditional pacemakers (the majority of which were dual-chamber). Important patient demographics included median age 66 years (IQR, 57-77 years) and 59% male.
The study investigators found that PoR occurred during nine MRI scans (3.5%) in eight patients. The clinical effect was a decrease in heart rate during MRI in four patients, and transient anomalous battery life indication in one patient. The authors reported that all devices functioned normally after MRI, and did not report any further clinical impact. All PoR events occurred in older generation devices (market release before 2002; implantation before 2005), and in one brand only (Medtronic). They reported that "Medtronic pacemakers implanted before 2005 had a 45% risk of PoR compared to no recorded incidents for newer Medtronic devices and devices made by other manufacturers." Over half of all devices in the study were Medtronic devices.
The investigators concluded that while their findings "suggest that most patients with CIEDs today can safely undergo MRI," it "should not be performed in pacemaker-dependent patients with older at-risk generators."
Hwang, Y. M., Kim, J., Lee, J. H., et al. (2016). Cardiac Implantable Electronic Device Safety during Magnetic Resonance Imaging. Korean Circ J, 46(6), 804-810. doi:10.4070/kcj.2016.46.6.804
The aim of this study was to evaluate the safety of conducting an MRI on patients with CIEDs in variable conditions, including cases with a previously known contraindication for this procedure.
This study used data from a single center retrospective collection of data from June 1992 to August 2015, evaluating the clinical outcomes and device parameter changes in patients with CIEDs who underwent an MRI at Asan Seoul, Korea. The cardiac devices were examined immediately before and after the MRI (within 48 hours), as well as during follow-up clinic visits from three to six months after the procedure. The investigators evaluated parameter changes by assessing battery voltage, pacing mode, lead capture thresholds, sensing signal amplitudes, and lead impedance. Additionally, clinical and device related information was acquired by chart review.
The analysis method included using frequencies for categorical, medians and inter-quartile ranges for continuous variables. Statistical analyses included paired t-test and an analysis of variance to compare continuous variables of the measured device’s parameters.
The study identified contraindications for MRI, but these patients were included in the study. Study defined contraindicated patients were those with (1) an abandoned lead, (2) epicardially located leads, (3) a scanning area in proximity to the device (such as thorax area), (4) devices implanted within the previous 6 weeks, or (5) individuals who were subjected to an MRI field strength >1.5 T.
The MRI protocol included: (1a) pacemaker settings were reprogrammed to a pacing-only mode and all atrial anti-tachycardia functions of the device were turned off during the procedure; (1b) implantable cardioverter-defibrillator settings were programmed to a pacing-only mode in pacemaker-dependent patients and ventricular anti-tachycardia pacing and low- and high-voltage shocks were turned off; (2) patient being monitored for at least 10 minutes with the pacemaker in the passive mode before entering the MRI scanner; (3) cardiologist being present throughout the entire MRI; (4) heart rate and oxygen saturation continuous monitoring with an ECG and a pulse oximeter; (5) audio contact between the patient and physicians so patients could inform the physician of any discomfort during the procedure; (6) following the MRI, devices were re-examined and reprogrammed to their original settings, including re-initiation of all anti-tachycardia functions. From June 1992 to March 2015, 40 patients (38 with a pacemaker and 2 with ICDs) underwent 50 MRIs at the site (34 at 1.5 T and 6 at 3.0 T) MRI. An MRI of the brain was the most frequently performed (21 patients, 25 MRIs), followed by a spine MRI (9 patients, 9 MRIs). Eleven patients had MR-conditional pacemakers and the other patients had MR-nonconditional devices.
Important patient demographics (sample size n=40) included median age of 64 years ranging from 17 to 83 years and 50% men. A total number of 40 patients with a CIED underwent 50 MRIs.
The study investigators found that twenty three patients had what the authors considered to be standard contraindications for MRI: (1) nonfunctional leads (n=1, 2.5%), (2) epicardially located leads (n=9, 22.5%), (3) scanning area in proximity to a device (n=9, 22.5%), (4) devices implanted within 6 weeks (n=2, 5%), and (5) MRI field strength at 3.0 Tesla (n=6, 15%).
All patients underwent a satisfactory MRI examination with no adverse events during or after the procedure. There were no significant changes in parameters or malfunctioning devices in any patients with CIEDs.
The investigators concluded that. the study demonstrates that MRI studies in patients with MR- nonconditional and MR-conditional devices is safe under close medical supervision during the examination and that patients with a standard contraindication to an MRI (58%) had no adverse events during the procedure or after the three month follow-up.
Kaasalainen, T., Pakarinen, S., Kivisto, S., et al. (2014). MRI with cardiac pacing devices - safety in clinical practice. Eur J Radiol, 83(8), 1387-1395. doi:10.1016/j.ejrad.2014.04.022
The aim of this study was provide a single center "real life" experience of performing MRI examinations in clinical practice on patients with cardiac pacemaker systems and evaluate the safety of using a dedicated safety protocol for these patients.
This study used data from a retrospective cohort of consecutive 68 MRI scans at 1.5T in 64 patients with pacing devices at a single center in Finland, between November 2011 and May 2013. The study evaluated a safety protocol by comparing the measured device parameters prior to and after MRI examinations. Atrial and ventricular pacing capture thresholds, lead impedances, P/R wave sensing amplitudes, and battery voltage were measured before, immediately after, and one month after MRI scanning.
The analysis method included summaries of absolute changes and percentages of change from the baseline parameters using medians and interquartile ranges (IQRs). Discrete variables were summarized as absolute numbers and percentages. Paired data were used to compare the pre- and post-scan samples, and the related-samples, while Wilcoxon signed-rank test, with MRI as the unit of analysis, compared the pacemaker variables. Non-normally distributed unpaired data were compared with independent-samples Mann–Whitney U test.
Key exclusions were the presence of abandoned or non-fixated leads. Additionally, when the pacing device was manufactured before 2000, MRI was only seldom performed.
The MRI protocol included: (1) pre-MRI examination where a cardiologist recorded device parameters, especially lead impedances and capture thresholds, sensing signal amplitudes, and battery voltage; (2a) for non-pacemaker-dependent patients, pacing mode was programmed to monitor-only; (2b) pacing mode was programmed to asynchronous for patients with no stable intrinsic rhythm; (2c) ICDs were programmed to therapy-off mode; (1d) MR-conditional systems were programed according to the instructions of the pacing device manufacturers; (2) prior to the MRI, radiographers checked the EMR system to ensure that the patient had visited the pacemaker policlinic and that the pacemaker was programmed for the MRI; (3) resuscitation equipment being available outside the MRI room during all examinations in case of an emergency; (4) electrocardiographic and pulse oximetry monitoring during MRI to detect any changes in heart rate or rhythm related to MRI-induced pacemaker inhibition, loss of pacemaker capture, or ventricular arrhythmias; (5) patients were monitored via a camera and asked to inform the investigators via an intercom f any torque or heating sensation, pain, palpitations or any other unusual symptoms during imaging; (6) devices were interrogated and reprogrammed to the original settings immediately after the examination by a cardiologist in either the Radiology Department or the pacemaker policlinic.
Important patient demographics (sample size n=64 patients with 68 scans) included mean age was 67 ± 14 years and 58% were men.
Of the 68 scans, 21 (31%) were of the thorax area, and 20 (29%), 17 (25%) and 16 (24%) of the examinations were MRI scans of spine, head and cardiac, respectively. The remainder were scans of the pelvis, liver, vagina, rectum, wrist, lung, carotid artery, soft tissue of the neck, pancreas and knee. Sixty (60) patients had a PM (including 22 (37%) MR-conditional and 38 (63%) MR-unsafe PMs), while two patients (3%) had an MR-unsafe CRT device and two (3%) had an MR-unsafe ICD system.
The study investigators found that all MRI examinations were completed safely. Two patients with an MR-unsafe pacemaker experienced a change in pacing rate when entering the MRI environment, in one patient the pacing rate rose from 70 to 100 bpm because the magnet-mode was unintentionally left active. During the scans, there were no unexpected changes in the heart rate or rhythm, shocks delivered, or sustained atrial or ventricular arrhythmias, torque or heating sensations, palpitations, pain, dizziness or other unusual symptoms during MRI.
All devices were interrogated after MRI, and no changes in the programmed parameters or any damage to the pacemaker circuits or movement of the pulse generator was observed.
There were no significant differences in the variable changes between the MR-conditional and MR-nonconditional pacing systems, or between scans of the thorax and other scan areas. For most of the participants, the distributions of the immediate and one-month changes in the device parameters were within the 20% of baseline values (the prespecified safe range), although some changes approached clinically important thresholds.
The investigators concluded that, when proper pacing device programming and patient monitoring was adhered to MRI examinations. There were no observed differences between the results of the MR-conditional and MR-unsafe devices and none between scans of the thorax area and of other scanning regions.
Mason, S., Osborn, J. S., Dhar, R., et al. (2017). Real world MRI experience with non-conditional and conditional cardiac rhythm devices after magnaSafe. J Cardiovasc Electrophysiol. doi:10.1111/jce.13351
The aim of this study was to provide additional safety and demographic information supporting broader clinical application of MRI across patient and device categories.
This study used data from a single-site, prospective registry of consecutively scanned patients with CIEDs who underwent clinically indicated MRIs between February 2014 and August 2016. The primary study safety outcomes were death, or generator or lead failure. Secondary outcomes were a battery voltage loss of >0.04 V, a decrease in P wave voltage of >50%, a decrease in R wave voltage of >25%, a threshold increase of >0.5 V, and an impedance change of >50 Ω. Prespecified subgroup comparisons of interest included thoracic versus non-thoracic scans and conditional versus non-conditional CIED device scans. Intergroup comparisons were descriptive, given limited subgroup sizes. All devices were interrogated immediately post-scan, and pre-scan parameters were restored. If any significant changes in CIED parameters were observed, the CIED was rechecked at two to seven days, at three months, and at six months post-scan.
The analysis method was not specified.
Key exclusions were any pacemaker or ICD with an atrial or ventricular lead with a threshold greater than 2.5 volts was not scanned unless the MRI is critical or if the lead is not essential for cardiac health; any pacemaker or ICD with a battery life of less than one year was not scanned in MRI; any lead that has been in place less than 4 weeks will not be scanned in MRI.
The MRI protocol included: (1) that all devices were to undergo interrogation in the MRI suite outside the magnet room; (2a) for patients with asymptomatic intrinsic rhythms, the device was programmed to no pacing; (2b) for pacemaker patients with no or insufficient intrinsic rhythms or symptomatic bradycardia, the device was programmed to an asynchronous pacing mode at a nominal low resting heart rate; (2c) for ICD patients, all tachycardia therapy functions were disabled, and pacing was programmed similar to that of the pacemaker-only population; (3) a cardiologist or other qualified physician with appropriate training supervise the study and that ACLS trained personnel; (4) a "crash cart", including a non-MRI compatible defibrillator and a transcutaneous pacemaker, be immediately available; (5) during the scan, all patients were monitored continuously for cardiac rhythm and hemodynamics (e.g., by continuous digital pulse blood pressure); (6) devices were interrogated immediately post-scan, and pre-scan parameters were restored.
Important patient demographics (sample size n=178 patients with 212 MRI scans) included mean age 66 ranging from 24 to 93 years and 57% men.
A total of 178 consecutive patients with CIEDs underwent 212 MRI scans which were all performed using a 1.5 T MRI with a limit of 2W/kg. Fifty-two (29%) were done on patients with ICDs; 111 (62%) on MR-nonconditional pacemakers patients, and 27 (13%) scans were done on MR-conditional pacemakers in 15 (8.4%). Devices with left ventricular leads were present in 17 (9.6%) and two subcutaneous ICDs were also were included unrestricted by scan site (i.e., including thoracic scanning) and device type (i.e., by manufacturer, by pacemaker vs ICD, by uni- versus biventricular, and by MRI-conditional vs. non-conditional). Scan locations included 87 (41%) Cervical-spine/head/neck scans, 28 (13%) Thoracic spine/cardiac/shoulder (thoracic) scans, 69 (33%) Lumbar-spine/abdomen/pelvis scans, and 28 (13%) lower extremity scans.
The study investigators found that there were no primary or secondary outcome event regardless of device type (ICD, pacemaker, CRT, MR-conditional, MR-nonconditonal). There were no parameter changes or device complications. For pacing dependent patients, there were no disruptions to pacing during the scan.
The investigators concluded that this study validates and extends findings from that of the large but inclusion-restricted MagnaSafe Registry, profiles MRI scanning in CIED patients in general clinical practice, and argues against replacing non-conditional with conditional devices when MRI is performed in a carefully controlled environment.
Muehling, O. M., Wakili, R., Greif, M., et al. (2014). Immediate and 12 months follow up of function and lead integrity after cranial MRI in 356 patients with conventional cardiac pacemakers. J Cardiovasc Magn Reson, 16, 39. doi:10.1186/1532-429X-16-39
The aim of this study was to generate evidence "supporting the hypothesis that it is safe to scan patients with cardiac pacemakers in a 1.5 T MRI, if close supervision and monitoring as well as adequate pre- and post-scan programming is provided."
This study used data from patients enrolled between July 2004 and January 2012 at Ludwig-Maximilians-University Munich, Medical Faculty
The analysis method included paired Wilcoxon rank sum test with continuity correction for continuous variables, and the Kruskal–Wallis test for categorical data with comparison between pre- and follow-up scans was performed using ANOVA.
Key exclusions were any devices implanted for < 2 months prior to the MRI scan, devices with a battery status of "Beginning of life", patients with an epicardial pacing lead or a known or suspected lead fracture.
The MRI protocol included: (1) full interrogation of all device information and impedance, sensing and capture function are measured immediately before and after MRI and at every follow-up exam; (2a) for pacemaker dependent patients devices were programmed to an asynchronous stimulation mode (2b) for non-pacemaker dependent patients the device was set to subthreshold pacing without changes to the sensing parameters in non-pacemaker dependent patients; (2c) magnet response, rate response, premature ventricular contraction response, noise response, ventricular sense response, conducted atrial fibrillation response, and tachyarrhythmia functions (monitoring, antitachycardia pacing) were disabled. (3) continuous monitoring was performed including telemetry, continuous pulse oximetry with plethysmographic waveform, and blood pressure measurements every three minutes; (4) a cardiologist was present for the entire scan; (5) resuscitation equipment was available in the MRI suite; (6) after the scan, and at every follow up patients were asked for clinical symptoms; (7) after the scan, each device was reprogrammed to its pre-scan settings.
Important patient demographics (sample size n=356) included mean age 61 ± 9 years and 64% men. A total of 356 patients with single (n = 132) or dual chamber (n = 224) cardiac pacemakers and urgent indication for a cranial MRI were followed regularly for 12 months after the scan.
The study investigators found that there were no induced arrhythmias, pacemaker dysfunction, or statistically significant changes in device program parameters. They reported that all devices were functioning appropriately after MRI, with the caveat that "although in 37 devices (10.4%) Power-on-Reset (PoR) occurred and in some reprogramming was necessary . . . ERI [elective replacement interval] was triggered and the ERI message could be cleared with the programmer to normal function after the scan."
The investigators concluded that study results supported the evidence that patients with conventional pacemakers can safely undergo cranial MRI at 1.5 T when a standard safety protocol is followed.
Nazarian, S., Hansford, R., Roguin, A., Goldsher, D., Zviman, M. M., Lardo, A. C., . . . Halperin, H. R. (2011). A prospective evaluation of a protocol for magnetic resonance imaging of patients with implanted cardiac devices. Ann Intern Med, 155(7), 415-424. doi:10.7326/0003-4819-155-7-201110040-00004
The aim of this study was define the safety of an MRI protocol for patients with a pacemaker or ICD, using device selection based on previous in vitro, in vivo, and pilot clinical studies, and device programming to minimize inappropriate activation or inhibition of therapies.
This study used data from a single-center predominate (94% at one US center), prospective, non-randomized trial was to evaluate the safety of a protocol for MRI at 1.5T in patients with a CIED enrolled consecutively between February 2003 and April 2010.
The analysis method included summarizing continuous variables as medians and interquartile ranges (IQRs) and discrete variables summarized as absolute numbers and percentages. Lead variables were compared by using the Wilcoxon signed-rank test with MRI as the unit of analysis.
Key exclusions were patients with newly implanted (<6 weeks) leads; those with abandoned or epicardial leads, and pacemaker-dependent patients with an ICD.
The MRI protocol included: (1) baseline and immediate follow-up interrogations were performed within minutes of MRI; (2) pacemaker dependence was assessed before MRI by transient inhibition of pacing; (3a) pacing mode was programmed to asynchronous for patients without a stable intrinsic rhythm; an inhibited pacing mode was used for other patients; (3b) all other pacing and tachyarrhythmia functions were disabled. After completion of MRI, devices were reprogrammed to original settings; (4) a registered nurse with experience in device programming and advanced cardiac life support was present during all scans, with immediate backup from a cardiac electrophysiologist; (5) during the scan patients were continuously monitored by using the MRI scanner in-room speaker system, noninvasive blood pressure measured (every 3 minutes), continuous electrocardiography, and pulse oximetry.
Important patient demographics (sample size n=438 patients with 555 MRI scans) included median age 66 years (IQR, 55 – 77 years) and 68% men. A total of 555 MRI scans were performed in 438 patients (54% with pacemakers and 46% with ICDs); 18% (or roughly 100 scans) were for thoracic MRIs (defined in this study as cardiac MRIs; 22% of scans were for spine MRIs and the number for thoracic spine MRIs was not reported).
"The pacing mode was changed to asynchronous for pacemaker-dependent patients and to demand for others. Tachyarrhythmia functions were disabled. Blood pressure, electrocardiography, oximetry, and symptoms were monitored by a nurse with experience in cardiac life support and device programming who had immediate backup from an electrophysiologist." Primary outcomes were episodes of activation or inhibition of pacing, patient symptoms, and changes in device settings (parameters).
The study investigators found that three of the 438 patients (receiving one cardiac, one brain, and one cervical spine MRI respectively) experienced an acute power-on-reset (POR) event during MRI scanning (up to 1.5% of device recipients). According to the authors, these POR episodes were "the primary clinically significant event attributable to MRI" in the trial. All three of these patients had devices implanted prior to the year 2000. None of these three patients had device dysfunction at long-term follow-up (463, 105, and 416 days, respectively). A total of 53 pacemaker-dependent patients without an ICD underwent MRI "without safety issues." For other device parameters, "significant variability was noted, and some changes approached clinically important thresholds."
The investigators concluded that MRI could be done safely in patients with selected cardiac devices when following an appropriate safety protocol. They noted that "because changes in device variables and programming may occur, electrophysiologic monitoring during MRI is essential." They also concluded that "the decision to perform MRI in each patient with an implantable device should be made by balancing the potential benefit of MRI against the attendant risks. Because thoracic MRI sequences have a greater effect on device variables and are more likely to result in artifacts, these sequences should be reserved for patients with an absolute clinical need."
Ono, M., Suzuki, M., & Isobe, M. (2017). Feasibility, safety, and potential demand of emergent brain magnetic resonance imaging of patients with cardiac implantable electronic devices. J Arrhythm, 33(5), 455-458. doi:10.1016/j.joa.2017.01.002
The aim of this study was to compare feasibility and safety of emergent and scheduled MRI orders for patients with MRI-conditional CIED.
This study used data from a single-center, retrospective analysis comparing emergent and scheduled MRI orders for patients with MR-conditional cardiac implantable electronic devices (ICDs and pacemakers) at Kameda Medical Center in Japan. Patients scanned from October 2012 to September 2016 were included. The study sought to compare the safety of emergent to scheduled MRIs.
The analysis method included a Mann–Whitney U test, Chi-squared test, and Fisher's exact test.
Key exclusions from the study were not detailed.
The MRI protocol included: (1) information of the patient and device were screened and confirmed by either the cardiologist or electrophysiologist as compatible with MRI; (2) a baseline interrogation to record the values, such as pacing threshold and lead impedance, and a change of settings to an MRI-compatible mode were conducted by clinical engineers; (3) during the scan patients were continuously monitored by oxygen saturation and electrocardiography; (4) equipment for advanced cardiac life support was available during the scanning; (5) during day-time hours, either the cardiologist or electrophysiologist in charge that day and all the related allied professionals were called for either the emergent or scheduled scanning and during night-time hours, the cardiologist and radiographers on call and staying in the hospital were called and clinical engineers in charge that night were recalled from their homes for the emergent scanning; (6) post-MRI, device settings were reprogrammed to the original state.
Important patient demographics (sample size n=57 MRI order) included 63% men with a mean age 81.1 ± 10.4 (emergent MRI) and 76.1 ± 6.1 (scheduled MRI).
A total of 11 emergent, 38 scheduled, and 8 unscheduled/ urgent MRI orders were identified. All emergent MRI orders were of patients with pacemakers while 35/38 scheduled MRIs were of patients with pacemakers. The majority of the scans were of the brain (10/11 emergent and 14/38 scheduled) and for the purpose of stroke evaluation (10/11 emergent 8/38 scheduled).
The study investigators found that nine out of the ten patients with an emergent MRI underwent successful emergent brain MRI. The one patient who could not undergo scanning was solely due to staffing shortage, but that patient received an MRI later that same day. All emergent MRI scans were completed safely with no complications.
The investigators concluded that, when precautions to safely conduct MRI were taken, it was feasible to perform 24-hr emergent MRI of patients with CIEDs.
Russo, R. J., Costa, H. S., Silva, P. D., et al. (2017). Assessing the Risks Associated with MRI in Patients with a Pacemaker or Defibrillator. N Engl J Med, 376(8), 755-764. doi:10.1056/NEJMoa1603265
The aim of this study was to determine the frequency of cardiac device–related clinical events and device setting changes among patients with non–MRI-conditional devices who undergo nonthoracic MRI at a magnetic field strength of 1.5 T, as well as to define a simplified protocol for screening, monitoring, and device programming for such patients.
This study used data from.a multicenter prospective, cohort embedded within the MagnaSafe Registry. Pacemaker-dependent patients with an ICD and patients with a CRT device were excluded. Primary outcomes were death from any cause, generator or lead failure, induced arrhythmia, loss of capture, or electrical reset during the scanning. Secondary outcomes involved changes in device settings.
The analysis method included separate analyses for the pacemaker and ICD cohorts. The Wilson score method without continuity correction was used to calculate 95% confidence intervals for single proportions for primary endpoint events. The linear association between lead age and each of the secondary end points was assessed with Pearson’s product moment correlation coefficient.
Key exclusions were: patients with intraorbital, intraocular retained metal fragments, intracranial vascular clips/ coils etc; ICD or pacemaker generator placement before 2002; patients with an ICD and pacing dependent; pregnancy; device generator battery voltage at elective replacement indicated; presence of abandoned leads (with the exception of post coronary artery bypass graft temporary epicardial pacing wires); presence of implanted cardiac device in the abdominal position.
The MRI protocol included: (1) pre-scan interrogation was conducted and baseline device parameter settings were noted; (2) devices were programmed into the appropriate modes (see figure below); (3) personnel trained in and equipment/ supplies needed to perform advanced cardiac life support (including a transcutaneous pacemaker) were available; (4) during the scan, patients were continuously monitored by blood pressure, pulse oximetry, cardiac rhythm, and patients were monitored (visualized and heard during the procedure); (5) if a medical professional other than a qualified physician monitored the procedure, a qualified physician directly supervised the key portions of the procedure (initial interrogation and postscan reprogramming) and furnished assistance and direction throughout the performance of the procedure.
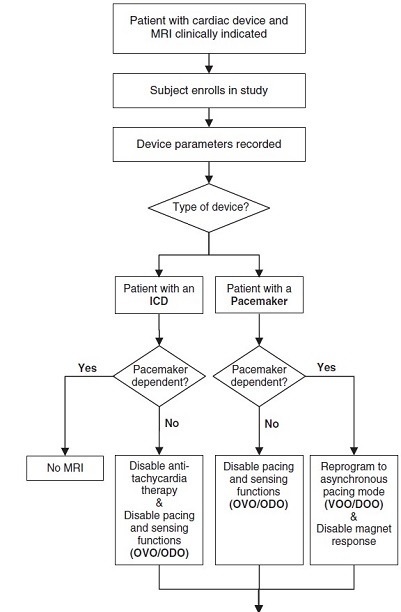
MRI was performed in 1000 cases in 818 patients with a pacemaker and 500 cases in 428 patients with an ICD (some patients had more than one MRI scan), from April 2009 through April 2014 at 19 centers throughout the United States. Important patient demographics (sample size n=1500 cases 1,000 pacemaker and 500 ICD) included mean age 73 ± 14 years, 58% male in the pacemaker group, and 65 ± 13 years, 69% male in the ICD group.
The study investigators found that no deaths, device failures, generator or lead replacements, loss of capture, or ventricular arrhythmias occurred during MRI. One patient with an ICD had not been programmed prior to the MRI according to the safety protocol; the ICD could not be interrogated after MRI and thus required immediate replacement. There were six cases of self-terminating atrial fibrillation or atrial flutter, and six cases of partial electrical reset. Repeat MRI was not associated with an increase in adverse events.
The investigators concluded that, in this prospective cohort study of 1,500 cases (1,246 patients), there were no deaths or device or lead failure "in any patient with a non–MRI conditional pacemaker or ICD who underwent clinically indicated nonthoracic MRI at 1.5 T, was appropriately screened, and had the device reprogrammed in accordance with the prespecified protocol."
Schwitter, J., Gold, M. R., Al Fagih, A., Lee, S., et al. (2016). Image Quality of Cardiac Magnetic Resonance Imaging in Patients With an Implantable Cardioverter Defibrillator System Designed for the Magnetic Resonance Imaging Environment. Circ Cardiovasc Imaging, 9(5). doi:10.1161/CIRCIMAGING.115.004025
The aim of this study was to assess the safety and efficacy of patients implanted with the Evera-MRI MR-conditional ICD system and subjected to an MRI examination.
This study used data from a multicenter (42 sites), international, randomized clinical trial. This specific publication from the Evera-MRI trial reported on the image quality performance of the two most frequently cardiac MRI types of pulse sequences [steady-state free precession (SSFP) and fast-gradient-echo (FGE)] acquisitions performed on a 1.5T scanner of patients implanted with the Evera-MRI MR-conditional ICD system.
The analysis method included an image quality assessment using a 7-point scale (1–3: good quality, 4-5: moderate quality, 6–7: nondiagnostic) and measuring ICD- and lead-related artifact size.
Key exclusions and specific MRI protocol were not explicitly detailed.
Important patient demographics (sample size n=263: 175 MRI and 88 Control) included mean age 59.7±13.8 years in those with MRI scan data.
In the MRI group, 156 subjects underwent a predefined not clinically indicated MRI examination scheduled at 9 to 12 weeks post implant. Of the 156 patients, 152 subjects had scan data collected for assessing cardiac image quality and therefore are included in this analysis.
The study investigators found that good to moderate image quality was obtained in 53% and 74% of SSFP and FGE acquisitions, respectively, covering the left ventricle, and in 69% and 84%, respectively, covering the right ventricle.
The investigators concluded that FGE produces better quality and smaller ICD-related artifacts for cardiac MRI than SSFP in patients with an MRI-conditional ICD system. In these patients implanted with ICD systems designed for the MR environment, cardiac MRI can offer diagnostic information in most cases.
Shah, A. D., Patel, A. U., Knezevic, A., et al. (2017). Clinical Performance of Magnetic Resonance Imaging Conditional and Nonconditional Cardiac Implantable Electronic Devices. Pacing Clin Electrophysiol, 40(5), 467-475. doi:10.1111/pace.13060
The aim of this study was to compare risks associated with MRI in patients with non-MRI conditional and MRI conditional pacing and defibrillator systems with particular attention to clinically actionable outcomes.
This study used data from a prospective, single-center observational study of patients having a CIED who were undergoing medically indicated MRI study, between October 2012 and July 2015, at the Emory University Hospital, underwent scanning at 1.5 T, and had pre-and postscan lead characteristic changes, system integrity, and symptoms analyzed. The primary endpoints included unintended programming changes, device resets, inappropriate antitachycardia therapies, and premature termination of the scan. The secondary endpoint was symptoms that did not require termination of the scan.
The analysis method included a comparison of endpoints between patients with MR-conditional and MR-nonconditional devices. Statisitcal analyses used a paired, two-tailed t-test and Wilcoxon rank-sum test. Results were expressed as mean change (95% confidence intervals).
Key exclusions were system implant duration <6 weeks, abandoned leads, ICD pulse generator manufacturer date before 2000, or pacemaker pulse generator date before 1998. However, the investigation did not exclude dependent patients with an ICD or pacemaker or patients with epicardial pacemaker leads, and there was no specified battery voltage requirement, though patients who were at Elective Replacement Interval or End of Life were not included
The MRI protocol included: (1) formal, face-to-face evaluation with a cardiac electrophysiologist that included device interrogation, assessment of lead characteristics, and periprocedural programming planning; (2a) pacemaker-dependent patients were programmed in an asynchronous mode; (2b) all trigger pacing type function (e.g., ventricular sense response) were inactivated; (2c) antitachycardia pacing and defibrillation therapies were inactivated and leads were programmed to a bipolar configuration; (3) a physician with expertise in device management (typically an electrophysiology fellow) was present in the imaging suite during scans for any patient with pacemaker dependency; (4) post-scan, devices were interrogated, lead characteristics were recorded, and devices were reprogramed as needed.
Important patient demographics (sample size n=105) included mean age of 65 years and 59% men. A total of 113 MRI scans were performed on 105 patients, allowing pre- and post-scan analysis of 90 atrial leads, 110 right ventricular leads, and nine left ventricular leads. Scans were performed in 16 (14%) patients with MR-conditional pacemakers, 74 (66%) MR-nonconditional pacemakers, and 39 (35%) MR-nonconditional defibrillators. Among the MR nonconditional devices 39 were defibrillators and nine were cardiac resynchronization devices. The following scan locations included: brain (50), abdomen (32), lumbar spine (16), pelvis (15), cervical spine (11), thoracic spine (5), chest (5), cardiac (3), neck (3), foot (1), and knee (1).
The study investigators found that small, nonsignificant changes in lead characteristics following scanning, with no significant difference between conditional and nonconditional devices. None of the lead parameter changes required revision or programming changes. There were no device resets, failures, or premature scan terminations. Imaging was not significantly impaired and MR studies were considered diagnostic in all cases. Inclusion of the pulse generator in the field of view was noted to be a frequent cause of artifact.
The investigators concluded that 1.5 T MRI scanning in patients with MRI conditional and non-MRI conditional cardiac devices was performed with similar, low clinical risk.
Sheldon, S. H., Bunch, T. J., Cogert, G. A., et al. (2015). Multicenter study of the safety and effects of magnetic resonance imaging in patients with coronary sinus left ventricular pacing leads. Heart Rhythm, 12(2), 345-349. doi:10.1016/j.hrthm.2014.11.037
The aim of this study was to evaluate the safety of MRI in patients with non-MRI-conditional coronary sinus LV pacing leads as part of their CIED system. MRI in patients with LV leads that course through the coronary sinus could potentially be associated with venous or lead thermal damage, dysrhythmias, or lead dysfunction or dislodgment.
This study used data from prospective data collected were at Mayo Clinic, Rochester, Minnesota (between September2009 and October 2013); Oklahoma Heart Institute, Tulsa, Oklahoma (between 2005 and 2013); and Intermountain Medical Center, Murray, Utah (between July 2007 and May 2013) in non–pacemaker-dependent patients with a non-MRI-conditional CIED system undergoing a medically necessary MRI.
The analysis method included use of descriptive statistics mean ± SD for normally distributed continuous variables, median and range or interquartile range for not normally distributed continuous variables, or number and percentage for categorical variables. Parameters of interest were compared between groups using the Pearson χ2 test for categorical variables and 2-sample t test or Wilcoxon rank-sum test for continuous variables, as appropriate.
Key exclusions were pacemaker-dependent patients, < 18 years old, troponin T >0.03 ng/mL in the absence of renal dysfunction or creatinine kinase-MB > 6.2 ng/mL, requiring general anesthesia for MRI, requiring continuous intravenous medication during MRI, and patients with abnormal device function (high pacing threshold; high pacing lead impedance; low battery voltage;and/or battery longevity prediction of <6 months or at the elective replacement indicator).
The MRI protocol included: (1) full device interrogation and reprogramming immediately before and after the MRI at the MRI site; (2a) devices were programmed to inhibited or a synchronous pacing mode; (2b) in the presence of symptomatic bradycardia, devices were programmed asynchronously to a rate faster than the intrinsic rate to avoid symptoms during the scan ICD tachyarrhythmia detection was programmed off; (3) post-MRI, devices were reprogrammed to their original settings; (3) Advanced Cardiac Life Support (ACLS)-trained pacing nurse (or cardiologist), technician, radiologist, and physicist supervised the scan; (4) an emergency cart including defibrillator was available in the event of cardiopulmonary compromise during the study; (5) patients were monitored using continuous vital sign, pulse oximetry, and ECG monitoring; (6) during the scan, visual and voice contact were maintained with the patient to monitor pain, discomfort, or other perceived abnormality.
Important patient demographics (sample size n=40 patients with 42 MRI scans) included a mean of age 67 ± 9 years and 60% male.
The study investigators found that no adverse events, including "no overall differences in pre- and post-MRI interrogation LV lead sensing, impedance, or threshold. No individual LV lead changes required intervention."
The investigators concluded that "MRI scanning was performed safely in non–pace-maker-dependent patients with coronary sinus LV leads who were carefully monitored during imaging without clinically significant adverse effect on LV lead function."
Strom, J. B., Whelan, J. B., Shen, C., et al. (2017). Safety and utility of magnetic resonance imaging in patients with cardiac implantable electronic devices. Heart Rhythm, 14(8), 1138-1144. doi:10.1016/j.hrthm.2017.03.039
The aim of this study was to determine: (1) major and minor adverse events in patients with non–MRI-conditional CIEDs undergoing MRI with a safety protocol; and (2) whether MRI results changed clinical management.
This study used data from a single-center, prospective cohort study at Beth Israel Deaconess Medical Center, Boston, MA with patients prospectively enrolled between June 19, 2014, and October 19, 2016. Major adverse events included loss of pacing, inappropriate shock or antitachycardia pacing, need for system revision, or death. Minor adverse events included inappropriate pacing, arrhythmias, power-on-reset events, heating at the generator site, or changes in device parameters at baseline or at 6 months.
The analysis method included categorical data being expressed as frequencies and percentages and continuous data as means and SDs. Adverse event proportions were presented with 95% confidence intervals (CIs) determined using the exact method based on binomial distributions.
Linear mixed-effects models were used to determine the mean difference, accounting for correlation of repeated outcomes within a given individual.
Key exclusions included patients with devices implanted <6 weeks, capped or abandoned leads or nontransvenous epicardial leads (exceptions made on case-by-case basis), and devices implanted before the year 2000.
The MRI protocol included: (1) prior to the scan, Electrophysiologist performed a full device interrogation to confirm current battery and lead parameters, and to save pre-MRI programmed settings; (2a) for patients with ICDs, all tachycardia detection and therapy was turned off; (2b) for both ICDs and PMs, PM-dependent patients were placed in an asynchronous pacing mode with pacing output changed to 5 V amplitude; (2c) nondependent patients were placed in demand mode and any additional features (rate response and ventricular sense response) that could impact pacing therapy were disabled; (3) all patients were monitored using wireless electrocardiographic telemetry as well as pulse oximetry with concurrent voice contact during the MRI; (4) post-MRI, devices were re-interrogated, battery and lead parameters checked, and original settings restored. A 1.5 T magnet was used for all studies.
Important patient demographics (sample size n=123 patients with 189 MRI scans) included mean/median age 70 ± 18.5 years (61.9% Medicare beneficiaries) and 63% men.
The study investigators found that there was only one major adverse event: one patient with loss of pacing (overall rate 0.5%). The rate of minor adverse events was also low (1.6%). Nearly all MR studies (98.4%) were interpretable, while 74.9% were determined to change clinical management according to prespecified criteria.
The investigators concluded that indicated MRI in patients with non-MRI-conditional ICDs performed with a safety protocol was safe and provided interpretable imaging that frequently influenced clinical management.
Taylor, A. J., Ellims, A., Lew, P. J., et al. (2013). Impact of cardiac magnetic resonance imaging on cardiac device and surgical therapy: a prospective study. Int J Cardiovasc Imaging, 29(4), 855-864. doi:10.1007/s10554-012-0131-4
The aim of this study was to evaluate the clinical utility of cardiac MRI in selecting patients for cardiac device implantation and/or cardiac surgery.
This study used data from a single-center, prospective study of all patients referred to the Alfred Hospital, Melbourne, Australia for clinical Cardiac magnetic resonance imaging (cMRI) scanning between July 1, 2007 and June 30, 2009.
The analysis method included use of mean ± standard deviation for continuous data and median ± interquartile range for ordinal data. Comparisons between multiple groups were made using either one way analysis of variance for continuous variables or Kruskal–Wallis one way ANOVA on ranks for ordinal variables with post hoc testing with the Holm Sidak method or Dunn’s method, respectively. Comparisons of proportions of multiple groups were made with multiple Chi-squared analyses implementing a Bonferroni correction.
Key exclusions and specific MRI protocol were not explicitly detailed.
Important patient demographics (sample size n=732) included a median age 49 ± 17 years and 66% men. A total of 732 patients received clinically indicated cMRI scans and there was six month follow-up data available for 666 of these patients. Of these 666 patients, 110 (17%) had preexisting CIEDs (72 with ICDs, 33 with CRT-D, 5 with pacemakers). Baseline data prior to the scan included planned cardiac device implantation and/or cardiac surgery. The primary outcome was the number of cardiac devices or surgical procedures averted that could be directly attributed to the cMRI results (and assuming 100% delivery of planned interventions if cMRI were not performed). Adverse events other than for death were not reported.
The study investigators found that on 6 month follow-up after cMRI, 56/150 (37%) of patients with an initial plan for device implantation or cardiac surgery did not undergo the planned intervention (P<0.001), while 33/516 (6%) of patients without an initial device or surgical plan ended up receiving device implantation or cardiac surgery as a result of the cMRI (P<0.001). Subgroup analysis of patients with pre-existing CIEDs was not reported.
The investigators concluded that cMRI significantly impacts clinical management with respect to patient selection for device implantation or surgery for cardiac disease.
Yadava, M., Nugent, M., Krebsbach, A., et al. (2017). Magnetic resonance imaging in patients with cardiac implantable electronic devices: a single-center prospective study. J Interv Card Electrophysiol. doi:10.1007/s10840-017-0262-6
The aim of this study was to determine the safety ofMRI in patients with CIEDs, using a protocol for patient selection and device programming.
This study used data from a prospective, single-center study conducted at Oregon Health and Science University which included patients with a MR-conditional and MR-nonconditional pacemakers or ICD and a clinical indication for MRI were enrolled from September 2012 to September 2015. The study assessed the safety of MRI in patients with CIEDs which were interrogated pre- and post-scan, and at follow-up one to six weeks later. All subjects underwent imaging with a 1.5 T scanner with no limitations placed on positioning of the magnetic isocenter, and no restriction on specific absorption rate.
The analysis method included continuous variables being described as mean and standard deviation, except for non-normally distributed variables, which are reported as median and interquartile range. The categorical and binary variables were summarized as counts and percentages. Comparisons of normally distributed variables between device groups was performed with two sample t-tests and non-normally distributed variables were compared with two-sample Wilcoxon tests. Categorical variables were tested for association with device with chi-square tests.
Key exclusions were those with a newly implanted device (<4 weeks), PPMs manufactured before 1996 and ICDs before 2000, those with epicardial and abandoned leads, and ICD patients with pacemaker-dependence, and pregnant patients in their first trimester (no gadolinium-enhanced scans in pregnant patients).
The MRI protocol included: (1) all devices were interrogated immediately prior to MRI; (2) pacemakers were programmed to an asynchronous mode if pacemaker-dependent and left in their baseline mode in patients without pacemaker-dependence, while tachycardia detection and therapies were disabled in eligible ICD patients; (3) all scans were done under the supervision of an electrophysiology nurse or physician assistant (PA) with experience in the implanted device functioning and advanced cardiac life support; (4) a cardiac electrophysiologist was on backup, and resuscitation equipment was readily available; (5) non-invasive monitoring of heart rhythm and hemodynamic variables was done for all patients; (6) patients communicated with the nurse or PA and MR technologist using a two-way microphone system; (7) post-MRI, devices were interrogated and re-programmed to their original settings.
Important patient demographics (sample size n=227 patients with 293 scans) such as age and gender were not provided. Devices included 170 (70.6%) pacemakers and 71 (29.5%) ICDs. One hundred ninety (83.7%) patients underwent a single scan. Thirty-seven (16.3%) had repeat scans. One hundred seventeen (39.9%) of the total scans were considered thoracic (chest; heart; thoracic spine).
Of the scanned patients, 27 (11.2%) had single-chamber pacemakers 136 (56.4%) dual-chamber pacemakers, 29 (12.0%) single-chamber ICDs, 21 (8.7%) dual-chamber ICDs, 7 (2.9%) biventricular pacemakers BiV, and 21 (8.7%) BiV ICDs. Of these, 12/170 (7.1%) pacemakers and 2/71 (2.8%) ICDs were MR-conditional.
The study investigators found that thirteen (4.4%) scans were aborted with six being due to image quality, six as a result of subjective complaints, and one terminated due to inappropriate device functioning which was due to MR induced noise being detected as AF by the device, resulting in intermittent mode-switching.
No scans had to be to be terminated due to overt device malfunction. Post-scan and follow-up interrogation demonstrated no changes in device parameters requiring reprogramming or revision.
The investigators concluded that, "apart from imaging artifact, the cause for scan termination was largely subjective complaints such as anxiety, claustrophobia, and pocket site symptoms. Whether this was due to a genuine effect of the MRI field on the generator versus a phantom phenomenon is unclear." They believe that the rate of non-completion due to subjective complaints 5/293 (1.7%) was similar to that reported in patients without CIEDs undergoing MR imaging.
4. Medicare Evidence Development & Coverage Advisory Committee (MEDCAC)
A MEDCAC meeting was not convened on this issue.
5. Evidence-Based Guidelines
There are no pertinent evidence-based guidelines.
6. Professional Society Recommendations / Consensus Statements / Other Expert Opinion
Expert Consensus Statement
Verma, A., Ha, A. C., Dennie, C., et al. (2014a). Canadian Heart Rhythm Society and Canadian Association of Radiologists consensus statement on magnetic resonance imaging with cardiac implantable electronic devices. Can Assoc Radiol J, 65(4), 290-300. doi:10.1016/j.carj.2014.08.001
Verma, A., Ha, A. C., Dennie, C., et al. (2014b). Canadian Heart Rhythm Society and Canadian Association of Radiologists consensus statement on magnetic resonance imaging with cardiac implantable electronic devices. Can J Cardiol, 30(10), 1131-1141. doi:10.1016/j.cjca.2014.07.010
The imaging facility should develop a standardized protocol to triage CIED patients for MR scanning. This protocol will systematically:
(1) Identify patients with CIED systems;
(2) Alert the MR team of the presence of a CIED in a given patient;
(3) Formalize a referral process to the CIED clinic to obtain information on the CIED and to assess its function;
(4) Identify potential relative contraindications that might increase risk during MR scanning;
(5) Ensure that the CIED and patient have been properly assessed in preparation for MR scanning;
(6) Ensure that the patient’s CIED is reinterrogated and reprogrammed after MR scanning; and
(7) Alert physicians (MR radiologist and CIED cardiologist) of potential CIED malfunction before, during, and afterMR scanning.
The authors note that MR scanning of patients with non-MR-conditional CIED systems is considered "off-label" and is not endorsed by regulatory agencies (e.g., Health Canada, US Food and Drug Administration), joint published guidelines from cardiovascular and radiology societies, and CIED manufacturers. As such, MR imaging of a patient with a non- MR-conditional CIED system is not routinely performed and is not considered to be standard of practice. However, the writing committee recognizes the existence of clinical scenarios in which MR scanning might provide crucial information in the management of the patient’s care. If this is the case, provisions can be made to allow for such "off-label" MR scanning to be performed with the understanding that serious and potentially life-threatening risks might occur.
The writing committee specifies that a detailed and explicit risk/benefit discussion be made among the: (1) referring physician (preferably a specialist in the specific body region of interest, such as a neurologist, neurosurgeon, orthopaedic surgeon, etc.); (2) cardiologist with expertise in CIED management; and (3) MR radiologist. The consensus recommendation of this group and the risks of "off-label" MR scanning must be documented and communicated to the patient or the patient’s substitute decision-maker. Written informed consent for MR scanning is requisite. Specifically, the following potential risks should be discussed:
(1) Pacemaker or ICD dysfunction;
(2) Pacemaker or ICD damage;
(3) Arrhythmia; and
(4) Death.
Recommendations:
- We recommend that MR imaging of MR-conditional CIEDs can be performed with a low risk of life threatening complications provided that patients and their CIEDs are properly evaluated before imaging and the scanning protocol be within the specified labelling for that CIED model (Strong Recommendation, Moderate-Quality Evidence).
- We recommend that facilities that perform MR scanning of patients with MR-conditional CIED systems should establish a formalized protocol via close collaboration between the CIED clinic and radiology department (Strong Recommendation, Low-Quality Evidence).
- We recommend that the specific roles for the CIED clinic prior to MR scanning of a patient with an MR-conditional CIED should include:
- Identification and confirmation of all elements of the CIED as MR-conditional;
- Evaluation of the CIED for potential functional abnormalities;
- Programming of the CIED to the appropriate MR imaging mode to avoid inappropriate pacing, device suppression, or inappropriate therapies (Strong Recommendation, Low-Quality Evidence).
- We recommend that the specific roles for the Radiology Department prior to MR scanning of a patient with an MR-conditional CIED should include:
- Triaging of MR requisitions to determine appropriateness of imaging;
- Initiation of pre-imaging preparation of the patient with the CIED clinic;
- Initiation of local standard operating imaging procedures to perform MR scanning in accordance to manufacturer- and radiologist-suggested parameters
(Strong Recommendation, Low-QualityEvidence).
- We recommend that during the MR scan, a member of the CIED clinic (technician, nurse, or physician) should be readily accessible (although not necessarily in person) to the MR imaging team for CIED management (Strong Recommendation, Low-Quality Evidence).
- We recommend that during the MR scan, the radiology suite must provide proper monitoring of CIED patients to minimize the occurrence of adverse events related to MR scanning. Basic monitoring requirements include methods for 2-way communication between operator and the patient and either pulse oximetry or telemetric ECG monitoring and access to emergency resuscitation equipment (Strong Recommendation, Low-Quality Evidence).
- We recommend that the patient be reassessed by the CIED clinic personnel to evaluate for CIED abnormalities after the MR scan and for the CIED to be reprogrammed to its original (prescan) settings (Strong Recommendation, Low-Quality Evidence).
- We recommend that a MR scan is contraindicated if any one or more of the following conditions exist:
- Suspected or known fractured pacing or ICD leads;
- Abandoned epicardial pacing or ICD lead(s) intended for permanent pacing or ICD therapy;
- Lead extenders, lead adaptors, or lead remnants that persist in the patient’s body (Strong Recommendation, Low-Quality Evidence).
- We recommend that MR imaging of a non-MR conditional CIED should only be performed at centres with a high level of expertise in MR imaging and CIED management. These centres must have established and well-defined imaging and vital status monitoring protocols, derived from close collaboration between the CIED clinic and radiology department (Strong Recommendation, Low-Quality Evidence).
Indik JH, Gimbel JR, Abe H, et al. 2017 HRS Expert Consensus Statement on Magnetic Resonance Imaging and Radiation Exposure in Patients with Cardiovascular Implantable Electronic Devices. Heart Rhythm 2017;14:e97-e153.
The HRS consensus statement was developed in collaboration with and endorsed by the American College of Cardiology (ACC), American College of Radiology (ACR), American Heart Association (AHA), American Society for Radiation Oncology (ASTRO), Asia Pacific Heart Rhythm Society (APHRS), European Heart Rhythm Association (EHRA), Japanese Heart Rhythm Society (JHRS), Pediatric and Congenital Electrophysiology Society (PACES), Brazilian Society of Cardiac Arrhythmias (SOBRAC), and Latin American Society of Cardiac Stimulation and Electrophysiology (SOLAECE) and in collaboration with the Council of Affiliated Regional Radiation Oncology Societies (CARROS).
The document cites that it was intended to help cardiologists, radiologists, radiation oncologists, and other health care professionals involved in the care of adult and pediatric patients with cardiac implantable electronic devices (CIEDs) who are to undergo magnetic resonance imaging (MRI), computed tomography, and/or radiation treatment. It provides an evidence review and recommendations regarding MRI scans in patients with MR conditional and MR nonconditional devices.
The Class of Recommendation (COR) indicates the strength of the recommendation and estimates the magnitude of benefit versus risk.
Class I (Strong):
- Is recommended.
- Should be performed/administered/other
Class IIa (Moderate):
- Is reasonable.
- Can be useful/effective/beneficial.
Class IIb (Weak):
- May/might be reasonable.
- May/might be considered
- Usefulness/effectiveness is unknown/unclear/uncertain or not well established.
Class III: No Benefit (Moderate):
- Is not recommended.
- Is not indicated/useful/effective/beneficial.
Class IV: Harm (Strong):
- Potentially harmful
- Causes harm
- Associated with excess morbidity/ mortality
- Should not be performed/administered/other
The Level of Evidence (LOE) rates the quality of the evidence based on the type, quantity, and consistency of the data from clinical trials and other sources.
Level A
- High-quality evidence from more than 1 RCT Meta-analyses of high quality RCTs
- One or more RCTs corroborated by high-quality registry studies
Level B-R
- Moderate-quality evidence from 1 or more RCTs
- Meta-analyses of moderate-quality RCTs
Level B-NR
- Moderate-quality evidence from 1 or more well-designed, well-executed nonrandomized studies, observational studies, or registry studies
- Meta-analysis of such studies
Level C-LD
- Randomized or nonrandomized observational or registry studies with limitations of design or execution Meta-analyses of such studies
- Physiological or mechanistic studies in human subjects
Level C-EO
- Consensus of expert opinion based on clinical experience
The following recommendations were put forward:
Management of Patients with a CIED Referred for MRI
MR Conditional Devices
Class I
- MR conditional devices should be considered MR conditional only when the product labeling is adhered to, which includes programming the appropriate "MR mode" and scanning with the prerequisites specified for the device. (LOE: A)
- MR imaging in a patient with an MR conditional system should always be performed in the context of a rigorously applied standardized institutional workflow, following the appropriate conditions of use. (LOE: B-R)
- It is recommended for patients with an MR conditional system that personnel with the skill to perform advanced cardiac life support, including expertise in the performance of CPR, arrhythmia recognition, defibrillation, and transcutaneous pacing, be in attendance with the patient for the duration of time the patient’s device is reprogrammed, until assessed and declared stable to return to unmonitored status. (LOE: B-R)
- It is recommended for patients with an MR conditional system that ECG and pulse oximetry monitoring be continued until baseline, or until other clinically appropriate CIED settings are restored. (LOE: A)
- All resuscitative efforts and emergency treatments that involve the use of a defibrillator/monitor, device programming system, or any other MRI unsafe equipment should be performed after moving the patient outside of Zone 4. (LOE: C-EO)
- It is recommended for patients with an MR conditional system that personnel with the skill to program the CIED be available as defined by the institutional protocol. (LOE: C-EO)
Class IIa
- It is reasonable to perform an MR scan on a patient with an MR conditional system implanted more recently than the exempt period for conditionality of the system, based on assessment of risk and benefit for that patient. (LOE: C-EO)
MR Nonconditional Devices
Recommendations for the Decision to Perform an MRI on Patients with an MR Nonconditional CIED
Class IIa
- It is reasonable for patients with an MR nonconditional CIED system to undergo MR imaging if there are no fractured, epicardial, or abandoned leads; the MRI is the best test for the condition; and there is an institutional protocol and a designated responsible MR physician and CIED physician. (LOE: B-NR)
- It is reasonable to perform an MR scan immediately after implantation of a lead or generator of an MR nonconditional CIED system if clinically warranted. (LOE: B-NR)
- For patients with an MR nonconditional CIED, it is reasonable to perform repeat MRI when required, without restriction regarding the minimum interval between imaging studies or the maximum number of studies performed. (LOE: C-LD)
Recommendations for the Management of Patients with an MR Nonconditional CIED Who Are to Have an MRI scan
Class I
- It is recommended for the patient with an MR nonconditional CIED that device evaluation be performed immediately pre- and post-MRI with documentation of pacing threshold(s), P- and R-wave amplitude, and lead impedance using a standardized protocol. (LOE: B-NR)
- A defibrillator/monitor (with external pacing function) and a manufacturer-specific device programming system should be immediately available in the holding area adjacent to the MR scanner room while an MR nonconditional CIED is reprogrammed for imaging. (LOE: B-NR)
- It is recommended that continuous MR conditional ECG and pulse oximetry monitoring be used while an MR nonconditional CIED is reprogrammed for imaging. (LOE: B-NR)
- It is recommended that personnel with the skill to perform advanced cardiac life support, including expertise in the performance of CPR, arrhythmia recognition, defibrillation, and transcutaneous pacing, accompany the patient with an MR nonconditional CIED for the duration of time the patient’s device is reprogrammed, until assessed and declared stable to return to unmonitored status. (LOE: B-NR)
- For patients with an MR nonconditional CIED who are pacing-dependent (PM or ICD), it is recommended that:
- Personnel with the skill to program the CIED be in attendance during MR scanning.
- A physician with the ability to establish temporary transvenous pacing be immediately available on the premises of the imaging facility.
- A physician with the ability to direct CIED programming be immediately available on the premises of the imaging facility. (LOE: B-NR)
- For patients with an MR nonconditional CIED who are not pacing-dependent, it is recommended that:
- Personnel with the skill to program the CIED be available on the premises of the imaging facility.
- A physician with the ability to direct CIED programming be available on the premises of the imaging facility. (LOE: B-NR)
- It is recommended that for the patient with an MR nonconditional CIED who is pacing-dependent to program their device to an asynchronous pacing mode with deactivation of advanced or adaptive features during the MRI examination, and the pacing rate should be selected to avoid competitive pacing. (LOE: B-NR)
- All tachyarrhythmia detections for patients with an ICD should be disabled prior to MRI. (LOE: B-NR)
- It is recommended that ECG and pulse oximetry monitoring be continued until baseline or until other clinically appropriate CIED settings are restored for patients with an MR nonconditional CIED. (LOE: C-EO)
- All resuscitative efforts and emergency treatments that involve the use of a defibrillator/monitor, device programming system, or any other MRI-unsafe equipment should be performed after moving the patient outside of Zone 4. (LOE: C-EO)
Class IIa
- For a patient with an MR nonconditional CIED who is not pacing-dependent, it is reasonable to program their device to either a nonpacing mode (OVO/ODO) or to an inhibited mode (DDI/VVI), with deactivation of advanced or adaptive features during the MRI examination. (LOE: B-NR)
- It is reasonable to programpatients with an MR nonconditional CRT device who are not pacing-dependent to an asynchronous pacing mode (VOO/DOO) with deactivation of advanced or adaptive features during the MRI examination, and with a pacing rate that avoids competitive pacing. (LOE: B-NR)
- For patients with an MR nonconditional CIED, it is reasonable to schedule a complete follow-up CIED evaluation within 1 week for a pacing lead threshold increase ≥1.0 V, P-wave or R-wave amplitude decrease ≥50%, pacing lead impedance change ≥50 Ω, and high-voltage (shock) lead impedance change ≥5 Ω, and then as clinically indicated. (LOE: C-EO)
7. Public Comment
Public comments sometimes cite the published clinical evidence and give CMS useful information. Public comments that give information on unpublished evidence such as the results of individual practitioners or patients are less rigorous and therefore less useful for making a coverage determination.
CMS uses the initial public comments to inform its proposed decision. CMS responds in detail to the public comments on a proposed decision when issuing the final decision memorandum. All comments that were submitted without personal health information may be viewed in their entirety by using the following link: https://www.cms.gov/medicare-coverage-database/details/nca-view-public-comments.aspx?NCAId=289&.
Initial Comment Period: 7/12/2017 – 8/11/2017
During the initial 30-day public comment period CMS received 17 comments, one of which could not be posted to the website due to it containing personal health information (PHI). We reviewed the comments in their entirety, including all referenced literature submitted.
The majority of comments were received from physicians, professional societies, and medical technology manufacturers. The remaining comments were from patients, a nurse, a health care provider and one individual who did not identify an affiliation or profession. Most of the comments mentioned the recently published MagnaSafe Registry as well as the Heart Rhythm Society (HRS) Expert Consensus Statement in support of revising the NCD to provide for patients with an MR non-conditional cardiovascular implantable electronic device (CIED). Several comments expressed that CMS’ intent for opening the reconsideration was unclear while others identified additional indications to be covered for MRI.
Below are a summary of the public comments that requested additional indications, outside of 220.2(C)(1) of the NCD Manual. We did not propose any coverage revisions outside of section 220.2(C)(1)(with corresponding changes for alignment in 220.2(B)(3). We note, with the exception of MRI during a viable pregnancy, that decisions on additional indications requested by commenters to be added to section 220.2 of the NCD Manual are made by local Medicare Administrative Contractors.
One commenter identified conditions and billing codes and suggested they should be covered. In the evidence submitted, which included four websites, to support the additional coverage indications, two of the articles supported screening indications which as explained below are outside the scope of this NCA. With regard to urethral diverticulum, while decisions are made by local Medicare Administrative Contractors, we note that an interested party may request a reconsideration of the NCD specifically for other indications (please see: https://www.cms.gov/Medicare/Coverage/DeterminationProcess/howtorequestanNCD.html)
We appreciated that an article was submitted with a public comment requesting that CMS add full body MRI for multiple mylema, monoclonal gammopathy of undetermined significance and solitary bone plasmacytoma. While not addressed in this proposed decision, as above, we note that an interested party may request a specific reconsideration.
Two commenters asked that we add MRI coverage for six-month follow-up for breast biopsy. We note that decisions on MRI for breast cancer diagnosis are currently made by the local Medicare Administrative Contractors and an interested party may request a specific NCD reconsideration.
We received a few comments asking that we add cancer screenings to this NCD. For example, one commenter requested full body MRI for cancer screenings. Section 220.2 of the NCD manual concerns coverage and non-coverage of diagnostic MRI test. Screening items and services are outside the scope of this NCD. For Medicare coverage of additional screening tests, specific statutory requirements must be met. These requirements include that the screening item or service must be: 1) reasonable and necessary for the prevention or early detection of illness or disability, 2) recommended with a grade of A or B by the United States Preventive Services Task Force and 3) appropriate for individuals entitled to benefits under Part A or enrolled under Part B.
We received one comment requesting that we remove preganancy as a contraindication. While not addressed in this proposed decision, as above, we note that an interested party may request a specific reconsideration.
VIII. CMS Analysis
National coverage determinations are determinations by the Secretary with respect to whether or not a particular item or service is covered nationally by Medicare (§1869(f)(1)(B) of the Act). In order to be covered by Medicare, an item or service must fall within one or more benefit categories contained within Part A or Part B, and must not be otherwise excluded from coverage. Moreover, with limited exceptions, the expenses incurred for items or services must be reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member (§1862(a)(1)(A) of the Act).
When making national coverage determinations, we evaluate the evidence related to our analytic questions based on the quality, strength and totality of evidence presented in the reviewed literature. As part of this evaluation, it is important to consider whether the evidence is relevant to the Medicare beneficiary population. In determining the generalizability of the results of the body of evidence to the Medicare population, we consider, at minimum, the age, race and gender of the study participants.
There are a number of structured methods for evaluating diagnostic tests. In past diagnostic imaging NCDs, we considered the evidence in the hierarchical framework of Fryback and Thornbury (1991) where Level 1 concerns technical quality of the images; Level 2 addresses diagnostic accuracy, sensitivity, and specificity of the test; Level 3 focuses on whether the information produces change in the physician's diagnostic thinking; Level 4 concerns the effect on the patient management plan; Level 5 measures the effect of the diagnostic information on patient outcomes; and Level 6 examine societal costs and benefits of a diagnostic imaging technology.
In our analysis, we generally look for sound evidence that shows the test is analytically and clinically valid (Levels 1-2) and that use of the test to guide treatment improves health outcomes (clinical utility, Levels 3-5).
Further, the current NCD (see section 220.2 of the NCD Manual) covers PMs and ICDs under CED. However, we propose to add cardiac resynchronization therapy pacemaker (CRT-P), or cardiac resynchronization therapy defibrillator (CRT-D) because these devices are considered combination pacemaker and defibrillator devices and because the majority of the evidence we reviewed included all four devices (i.e., PMs, ICDs, CRT-Ps, and CRT-Ds). In addition, the HRS consensus document also included these same devices. Therefore, based on the evidence reviewed we propose to include all of these devices within this proposed decision.
While a detailed discussion of MRI technology is beyond the scope of this decision, MRI is considered the gold standard for imaging patients with multiple sclerosis (MS) (Lohrke 2016, Polman 2011) to identify appropriate patients for beneficial treatments (Cochrane Reviews: La Mantia 2012, Tramacere 2015). MRI is also used in identifying candidates for coronary revascularization (Campbell 2014, Greenwood 2012, Jaarsma 2012) and is included in appropriate use of neuroimaging in the diagnostic workup of dementia (Health Quality Ontario 2014, Bermingham 2014). Concerns in patients with certain implanted cardiac devices have been reported and include pacing alterations, inappropriate ICD discharges, mechanical pull and rotation of the device have been reported (Schoenfeld, 2007).
For this reconsideration, CMS focused on the following questions:
Q1. Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications informs the diagnosis or clinical management decisions in patients with implanted PMs or ICDs (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators)?
Q2. Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications improves health outcomes in patients with implanted PMs or ICDs (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators)?
Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications informs the diagnosis or clinical management decisions in patients with implanted PMs or ICDs (or combination cardiac resynchronization therapy pacemakers or cardiac resynchronization therapy defibrillators)?
Yes. Overall, based on the preponderance of evidence including new clinical studies, we propose that the evidence is sufficient to conclude that MRI performed for clinically appropriate imaging indications inform the diagnosis and clinical management of patients with PMs, ICDs, CRT-Ps and CRT-Ds (also known as cardiovascular implanted electronic devices (CIEDs) in the published evidence). Since the validity (analytic and clinical validity; Fryback and Thornbury Levels 1-4) of MRI in general patient populations has been studied and reported (Health Quality Ontario 2014, La Mantia 2012, Lohrke 2016, Tramacere 2015), the consideration of this question focuses on the ability to obtain the same MRI image quality in the presence of CIEDs. A valid MRI scan is important to guide clinical decision making and subsequent treatment based upon the results. If image quality is unaffected by the presence of CIEDs, then it would be likely that MRI test parameters such accuracy, sensitivity and specificity would be maintained and comparable to patients that do not have CIEDs as previously reported.
Studies from Kaasalainen et al. 2014 (n=64), Schwitter et al. 2016 (n=263), Strom et al. 2017 (n=123) and Taylor et al. 2013 (n=732) found that MRI studies of patients with CIEDs with FDA labeling specific to use in an MRI environment and those without specific labeling were performed with an adequate image quality for diagnosis. These diagnosis frequently directed clinical management including change in therapies. Observational studies by Strom et al. and Taylor et al. showed that MRI results in patients with CIEDs provided interpretable images that influenced clinical management. Given the maintenance of image quality and the published studies that reported changes in patient management, we believe the evidence is sufficient. We believe the proposed NCD reflects the current evidence in the peer-reviewed medical literature and professional society consensus statements.
Is there adequate evidence to conclude that MRI performed for clinically appropriate imaging indications improves health outcomes in patients with implanted PMs or ICDs?
Yes. Overall, based on the published evidence including new clinical studies and evidence-based guidelines, we believe that the evidence is sufficient to conclude that MRI performed for clinically appropriate imaging indications improves health outcomes in patients with implanted PMs or ICDs, CRT-P and CRT-D (hereinafter referred to as CIED). After finding that MRI in patients with CIEDs is comparably valid (analytically and clinically) to patients without CIED and it directed patient management, we assessed whether meaningful benefits outweighed harms to improve health outcomes (clinical utility; Fryback and Thornbury Level 5).
We analyzed published studies to determine whether the presence of a CIED increases harms assuming benefits of MRI are the same as patients without CIEDs. In a randomized trial (n=263) providing generally good evidence, Gold and colleagues reported that "no adverse effects were noted with a standardized, comprehensive MRI protocol" and that "pacing and sensing were not significantly affected by MRI." Large observational studies by Nazarian et al. (2011)(n=438) and Russo et al. (2017)(n=1246) showed that patients with CIEDs who underwent MRI did not experience harms such as deaths or device failures. These studies provide supporting evidence in broader populations. A number of smaller observational studies also showed consistent results. Since the published evidence showed comparable image quality and did not show an increase in harms of MRI in patients with CIEDs, there is sufficient evidence that MRI improves outcomes similar to patients without CIEDs.
In order to protect patients and to ensure these diagnostic tests are reasonable and necessary under Section 1862(a)(1)(A), we are proposing the requirements for MRI scans that do not have FDA labeling specific for patients with a CIED for use in an MRI environment. We are proposing these criteria based on the evidence and studies reviewed which used similar safety protocols for MRIs for patients with CIEDs. Further the HRS consensus document also recommends these safety criteria should be implemented. These safety criteria are similar to the criteria FDA requires in the label for all on label indications. We note that the criteria listed below do not apply to the scans that are done within the FDA label because it would be a duplicative requirement.
The following criteria which are based on our evidentiary review, including the HRS consensus guidelines, are proposed for any MRI scan for patients with a CIED but do not have a FDA label specific for this use in an MRI environment:
Tesla: Tesla (T) is a unit of measure of the strength of the magnetic field. Most MRI scanners are either 1.5T or 3.0T, with higher strength machines reportedly providing better images in less time but at higher cost. The preponderance of the reviewed evidence studied CIEDs within the 1.5 T MRI scan environment. Hwang et al. (2016) assessed outcomes in patients scanned with 1.5 T and 3.0 T MRI scanners. While Hwang et al. (2016) reported no clinically significant changes to device parameters or adverse events, this study reported on a limited patient experience (N=6) with respect to exposure to 3.0 T MRI. There is thus a paucity of evidence to support that the benefits of using 3.0 T MRI in patients with CIEDs that do not have FDA labeling specific to use in an MRI environment outweigh the harms. We also believe that such patients with a need for MRI will almost always have access to a 1.5 T MRI if indeed they have access to a 3.0 T MRI. Therefore, we propose to cover our Medicare beneficiaries who have a CIED without FDA-approved labeling for use in an MRI environment only for MRI scans at 1.5 T or below.
Post CIED Implant Waiting Period ≥ 6 week: Almost all studies reviewed excluded patients with recently implanted, revised, or modified leads. Investigators stated that this exclusion was due to lead dislodgements being more likely to occur in the immediate post-implantation period. The Canadian Heart Rhythm Society and Canadian Association of Radiologists consensus statement considers a recent CIED implant to be "red flags" for a CIED patient who is scheduled for MR scanning. As stated by that consensus statement and the HRS consensus statement, some CIED manufacturers recommend that a device with FDA labeling specific to use in an MRI environment be implanted > 6 weeks from time of MR imaging and a 6-week waiting period was adopted in clinical trials of PMs with FDA labeling specific to use in an MRI environment to avoid confusion as to whether a lead dysfunction was related to performance of the MRI scan. Only a few studies, including Nazarian, S (2011) and Friedman, H.L. (2013), provided limited observations regarding patients with MRI scans < 42 days after CIED implant. While these studies reported no clinically significant differences in device function observed between patients scanned early or late after CIED implantation, the subsets with earlier scans were very small. Therefore, we propose to cover Medicare beneficiaries with a CIED without FDA-approved labeling for use in an MRI environment only when beneficiaries are ≥ 6 weeks since CIED implantation or any lead revision or surgical modification.
Pacemaker-Dependent Patients: Electromagnetic interference (EMI) generated by the gradient magnetic field during MRI may be received by a CIED as a reset signal (Power on Reset, or PoR). This PoR could cause the CIED to revert to its factory default settings. For pacemaker-dependent patients with CIEDs programmed for asynchronous pacing used during MRI, the device may be reset to an inhibited mode. The evidence base, including studies by Higgins, J.V., et al. 2015 and Muehling, O.M., et al. 2014, observed occurrences of PoR which were at times associated with a decrease in heart rate during MRI. All devices functioned normally after completion of the MRI. Therefore, we are proposing to not include pacemaker-dependent patients under the covered population.
Fractured, Epicardial, or Abandoned Leads: The HRS consensus statement concluded that, "At the present time, however, there are insufficient data to comment on the safety of MRI performance with abandoned, epicardial, or fractured leads. The Canadian Heart Rhythm Society and Canadian Association of Radiologists consensus statement states that, "MR scanning is absolutely contraindicated" in the patients with fractured, epicardial, or abandoned leads. Postsurgical temporary epicardial leads that have been partially removed are not considered to be abandoned pacing leads." Patients with fractured, epicardial, or abandoned leads are frequently excluded from studies of CIEDs in the MRI environment. There were no MRI studies specifically on safety and outcomes of these patients which met our inclusion criteria. There is a paucity of evidence to support MRI scans in patients with fractured, epicardial, or abandoned leads. Therefore, we propose not to include patients with these lead conditions under the covered population for those with CIEDs.
Considerations in Patients with CIEDS during MRI: A review article (Schoenfeld, 2007) states that "…(p)otential interactions (of PMs) with MRI include pacing inhibition, inappropriate ICD discharges, rapid pacing, mechanical pull and rotation of the device, and device reprogramming," and suggests strategies to improve safety of MR scanning for patients with PMs and ICDs: "…Certain strategies to minimize complications have been suggested, including the use of less powerful MRI machines; imaging limited to extremities (i.e., remote from the implanted device); careful reprogramming of the intracardiac device, including asynchronous modes and maximal pacing output; selection of appropriate spin sequences; limitation of MRI to patients who are not pacemaker dependent; and careful, continuous peri-procedure monitoring."
Checklist: The published evidence base demonstrated that MRI scans in patients with CIED, when conducted under a checklist can be conducted without major adverse events. The HRS consensus statement highlighted the need for a standardized collaborative institutional policy which identifies personnel responsibilities and workflow, including assessment of the benefits of MR imaging compared with alternatives, protocols for pre- and post-scan CIED evaluation, and appropriate programming during the scan based on device and patient characteristics. The Canadian Heart Rhythm Society and Canadian Association of Radiologists consensus statement recommends that facilities performing MRI in patients with CIEDs that are not FDA labeled for use in an MRI environment should establish a formalized protocol via close collaboration between the CIED clinic and radiology department, to include properly evaluating patients and their CIED before and after imaging. Therefore, we propose to cover MRI scans for patients with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator but do not have a FDA label specific for this use in an MRI environment if the facility develops a checklist with the following criteria:
- patient assessment is performed to identify the presence of an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator.
- before the scan benefits and harms of the MRI scan are communicated with the patient or the patient’s delegated decision-maker;
- prior to the MRI scan, the implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator is interrogated and programmed into the appropriate MR scanning mode;
- a qualified physician, nurse practitioner or physician assistant with expertise with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator must directly supervise;
- a discharge plan that includes before being discharged from the hospital/facility, patient is evaluated and the an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator reinterrogated to detect and correct any abnormalities that might have developed during the MRI.
Coverage with Evidence Development (Coverage with Study Participation) Requirement:
In 2011, CMS posed questions regarding the evidence which CED studies to address (see Appendix C for the current 220.2 NCD). Based on our concerns at the time, we required additional data to be collected via study participation (see https://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=177).
We assessed the extent to which the published literature, including completed CED studies, addressed the following questions. (Each approved study had to address one or more aspects of one or more of the CED questions below.)
- Do results of MRI in PM/ICD beneficiaries with implanted cardiac devices affect physician decision making related to:
- Clinical management strategy (e.g., in oncology, toward palliative or curative care);
- Planning of treatment interventions; or
- Prevention of unneeded diagnostic studies or interventions, or preventable exposures?
- Do results of MRI in PM/ICD beneficiaries with implanted cardiac devices affect patient outcomes related to:
- Survival;
- Quality of life; or
- Adverse events during and after MR scanning?
Since the 2011 NCD, there have been nine approved clinical studies of MRI under CED. One of the nine studies has reached completion. Based on our analysis of the 18 reviewed publications in the Evidence section above, which includes the completed CED study, all 18 publications were directly related to at least one of the two CED questions. After reviewing the totality of this new evidence, we believe that the CED questions have been sufficiently answered and we believe that additional data collection is no longer needed.
We acknowledge that only one of the CED studies has been completed, and there are eight ongoing studies. However, the weight of the published literature in this field provides convincing evidence that, with appropriate precautions, MRI can be performed with minimal risk in Medicare beneficiaries with CIEDs, that the resulting images are of diagnostic quality, and that results of the MRI studies generally impact clinical management and improve patient health outcomes.
While we propose to end the CED requirement, we encourage the continuation and improvement of a voluntary registry for purposes of identifying strategies to further reduce the risk of minor complications and to develop device-specific MRI scanning guidance.
Considerations for Further Research:
The MRI studies reviewed implement protocols designed to minimize the risk of harm to patients with CIEDs who need an MRI. The large cohort studies by Russo (2017) of 1500 cases reported in the MagnaSafe Registry and Nazarian et al. 2011 of 555 cases provide strong evidence that appropriately performed and clinically indicated MRI is safe in patients with CIEDs. However, these and other studies highlight that the combination of pulse generator, lead type, lead positioning in the MRI system, and the magnitude of the electromagnetic field (EMF), can all affect the response of CIED systems to the EMF that is generated during MRI scanning (Delfino, J.G., Viohl, I, Woods, T.O., 2017).
While the studies reviewed demonstrated that there were no serious adverse events observed, they did not evaluate every potential generator/ lead combination and there were some rare, minor complications noted. A larger comprehensive registry of patients with CIEDs that do not have FDA labeling specific to use in an MRI environment undergoing MRI could be helpful moving forward to identify risks and suggest strategies to further reduce the risk of those minor complications and to develop implant-specific MRI scanning guidance. Such a registry could build off of the HRS "Checklist for MRI safety in the setting of implanted devices (PM or ICD)" detailed within their 2017 consensus statement, and include variables such as pulse generator and lead type, lead length, scan SAR, Tesla levels, and scan location.
Health Disparities
CMS is concerned about disparities in healthcare in the Medicare population, and when performing this assessment of the literature, there was little information addressing age, gender, race/ethnicity; socioeconomic status; or sexual orientation of study participants.
Summary
This NCA has focused on the contraindications for implanted pacemaker (PM), implantable cardioverter defibrillator (ICD), cardiac resynchronization therapy pacemaker (CRT-P), or cardiac resynchronization therapy defibrillator (CRT-D) in patients undergoing MRIs both on and off FDA label. Based on our analysis of the evidence published since the 2011 NCD, we propose to remove the contraindication in section 220.2(C)(1)(with corresponding changes for policy alignment in section 220.2(B)(3)) of the NCD Manual for Medicare coverage of MRI in beneficiaries with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator and provide specific conditions required for coverage. Furthermore, we propose removing the 2011 CED requirement. The evidence generated and reviewed since the 2011 NCD sufficiently answers the CED questions as noted above, and we believe that additional data collection is no longer needed for Medicare coverage purposes.
IX. Conclusion
We propose that the evidence is sufficient to conclude that magnetic resonance imaging (MRI) for Medicare beneficiaries with an implanted pacemaker (PM), implantable cardioverter defibrillator (ICD), cardiac resynchronization therapy pacemaker (CRT-P), or cardiac resynchronization therapy defibrillator (CRT-D) is reasonable and necessary for the diagnosis or treatment of illness or injury or to improve the functioning of a malformed body member under section 1862(a)(1)(A) of the Social Security Act under certain circumstances. Thus, we are proposing to modify our national coverage determination to eliminate the collection of additional information under the Coverage with Evidence Development paradigm under section 1862(a)(1)(E) of the Social Security Act.
We summarize these changes below and present our proposed changes fully in Appendix B. We will explain the proposed changes in the analysis section of this NCD decision memo. In general, we propose to:
- revise the language in section 220.2(C)(1) to remove the contraindication for Medicare coverage of MRI in a beneficiary who has an implanted pacemaker (PM) or implantable cardioverter defibrillator (ICD);
- expand coverage to include cardiac resynchronization therapy pacemaker (CRT-P), or cardiac resynchronization therapy defibrillator (CRT-D) devices;
- expand coverage for beneficiaries who have an implanted FDA-approved pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator correspondingly under 220.2(B)(3) of the NCD Manual as a Nationally Covered MRI indication;
- expand coverage for beneficiaries with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or
cardiac resynchronization therapy defibrillator device that do not have FDA labeling specific for an MRI with certain criteria;
- remove the Coverage with Evidence Development requirement.
See Appendix B for proposed manual language.
APPENDIX A
General Methodological Principles of Study Design
(Section VI of the Decision Memorandum)
When making national coverage determinations, CMS evaluates relevant clinical evidence to determine whether or not the evidence is of sufficient quality to support a finding that an item or service is reasonable and necessary. The overall objective for the critical appraisal of the evidence is to determine to what degree we are confident that: 1) the specific assessment questions can be answered conclusively; and 2) the intervention will improve health outcomes for patients.
We divide the assessment of clinical evidence into three stages: 1) the quality of the individual studies; 2) the generalizability of findings from individual studies to the Medicare population; and 3) overarching conclusions that can be drawn from the body of the evidence on the direction and magnitude of the intervention’s potential risks and benefits.
The methodological principles described below represent a broad discussion of the issues we consider when reviewing clinical evidence. However, it should be noted that each coverage determination has its unique methodological aspects.
Assessing Individual Studies
Methodologists have developed criteria to determine weaknesses and strengths of clinical research. Strength of evidence generally refers to: 1) the scientific validity underlying study findings regarding causal relationships between health care interventions and health outcomes; and 2) the reduction of bias. In general, some of the methodological attributes associated with stronger evidence include those listed below:
- Use of randomization (allocation of patients to either intervention or control group) in order to minimize bias.
- Use of contemporaneous control groups (rather than historical controls) in order to ensure comparability between the intervention and control groups.
- Prospective (rather than retrospective) studies to ensure a more thorough and systematical assessment of factors related to outcomes.
- Larger sample sizes in studies to demonstrate both statistically significant as well as clinically significant outcomes that can be extrapolated to the Medicare population. Sample size should be large enough to make chance an unlikely explanation for what was found.
- Masking (blinding) to ensure patients and investigators do not know to that group patients were assigned (intervention or control). This is important especially in subjective outcomes, such as pain or quality of life, where enthusiasm and psychological factors may lead to an improved perceived outcome by either the patient or assessor.
Regardless of whether the design of a study is a randomized controlled trial, a non-randomized controlled trial, a cohort study or a case-control study, the primary criterion for methodological strength or quality is to the extent that differences between intervention and control groups can be attributed to the intervention studied. This is known as internal validity. Various types of bias can undermine internal validity. These include:
- Different characteristics between patients participating and those theoretically eligible for study but not participating (selection bias).
- Co-interventions or provision of care apart from the intervention under evaluation (performance bias).
- Differential assessment of outcome (detection bias).
- Occurrence and reporting of patients who do not complete the study (attrition bias).
In principle, rankings of research design have been based on the ability of each study design category to minimize these biases. A randomized controlled trial minimizes systematic bias (in theory) by selecting a sample of participants from a particular population and allocating them randomly to the intervention and control groups. Thus, in general, randomized controlled studies have been typically assigned the greatest strength, followed by non-randomized clinical trials and controlled observational studies. The design, conduct and analysis of trials are important factors as well. For example, a well-designed and conducted observational study with a large sample size may provide stronger evidence than a poorly designed and conducted randomized controlled trial with a small sample size. The following is a representative list of study designs (some of that have alternative names) ranked from most to least methodologically rigorous in their potential ability to minimize systematic bias:
Randomized controlled trials
Non-randomized controlled trials
Prospective cohort studies
Retrospective case control studies
Cross-sectional studies
Surveillance studies (e. g. , using registries or surveys)
Consecutive case series
Single case reports
When there are merely associations but not causal relationships between a study’s variables and outcomes, it is important not to draw causal inferences. Confounding refers to independent variables that systematically vary with the causal variable. This distorts measurement of the outcome of interest because its effect size is mixed with the effects of other extraneous factors. For observational, and in some cases randomized controlled trials, the method in that confounding factors are handled (either through stratification or appropriate statistical modeling) are of particular concern. For example, in order to interpret and generalize conclusions to our population of Medicare patients, it may be necessary for studies to match or stratify their intervention and control groups by patient age or co-morbidities.
Methodological strength is, therefore, a multidimensional concept that relates to the design, implementation and analysis of a clinical study. In addition, thorough documentation of the conduct of the research, particularly study selection criteria, rate of attrition and process for data collection, is essential for CMS to adequately assess and consider the evidence.
Generalizability of Clinical Evidence to the Medicare Population
The applicability of the results of a study to other populations, settings, treatment regimens and outcomes assessed is known as external validity. Even well-designed and well-conducted trials may not supply the evidence needed if the results of a study are not applicable to the Medicare population. Evidence that provides accurate information about a population or setting not well represented in the Medicare program would be considered but would suffer from limited generalizability.
The extent to that the results of a trial are applicable to other circumstances is often a matter of judgment that depends on specific study characteristics, primarily the patient population studied (age, sex, severity of disease and presence of co-morbidities) and the care setting (primary to tertiary level of care, as well as the experience and specialization of the care provider). Additional relevant variables are treatment regimens (dosage, timing and route of administration), co-interventions or concomitant therapies, and type of outcome and length of follow-up.
The level of care and the experience of the providers in the study are other crucial elements in assessing a study’s external validity. Trial participants in an academic medical center may receive more or different attention than is typically available in non-tertiary settings. For example, an investigator’s lengthy and detailed explanations of the potential benefits of the intervention and/or the use of new equipment provided to the academic center by the study sponsor may raise doubts about the applicability of study findings to community practice.
Given the evidence available in the research literature, some degree of generalization about an intervention’s potential benefits and harms is invariably required in making coverage determinations for the Medicare population. Conditions that assist us in making reasonable generalizations are biologic plausibility, similarities between the populations studied and Medicare patients (age, sex, ethnicity and clinical presentation) and similarities of the intervention studied to those that would be routinely available in community practice.
A study’s selected outcomes are an important consideration in generalizing available clinical evidence to Medicare coverage determinations. One of the goals of our determination process is to assess health outcomes. These outcomes include resultant risks and benefits such as increased or decreased morbidity and mortality. In order to make this determination, it is often necessary to evaluate whether the strength of the evidence is adequate to draw conclusions about the direction and magnitude of each individual outcome relevant to the intervention under study. In addition, it is important that an intervention’s benefits are clinically significant and durable, rather than marginal or short-lived. Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits.
If key health outcomes have not been studied or the direction of clinical effect is inconclusive, we may also evaluate the strength and adequacy of indirect evidence linking intermediate or surrogate outcomes to our outcomes of interest.
Assessing the Relative Magnitude of Risks and Benefits
Generally, an intervention is not reasonable and necessary if its risks outweigh its benefits. Health outcomes are one of several considerations in determining whether an item or service is reasonable and necessary. CMS places greater emphasis on health outcomes actually experienced by patients, such as quality of life, functional status, duration of disability, morbidity and mortality, and less emphasis on outcomes that patients do not directly experience, such as intermediate outcomes, surrogate outcomes, and laboratory or radiographic responses. The direction, magnitude, and consistency of the risks and benefits across studies are also important considerations. Based on the analysis of the strength of the evidence, CMS assesses the relative magnitude of an intervention or technology’s benefits and risk of harm to Medicare beneficiaries.
APPENDIX B
Medicare National Coverage Determinations Manual
Draft
We are seeking public comments on the proposed language that we would include in the Medicare National Coverage Determinations Manual. This proposed language does not reflect public comments that will be received on the proposed decision memorandum, and which may be revised in response to those comments.
Table of Contents
(Rev.)220.2 - Magnetic Resonance Imaging (MRI)
(Rev., Issued: XX-XX-XX, Effective:XX-XX-XX,
Implementation:)
A. General
1. Method of Operation
Magnetic Resonance Imaging (MRI), formerly called nuclear magnetic resonance (NMR), is a non-invasive method of graphically representing the distribution of water and other hydrogen-rich molecules in the human body. In contrast to conventional radiographs or computed tomography (CT) scans, in which the image is produced by xray beam attenuation by an object, MRI is capable of producing images by several techniques. In fact, various combinations of MRI image production methods may be employed to emphasize particular characteristics of the tissue or body part being examined. The basic elements by which MRI produces an image are the density of hydrogen nuclei in the object being examined, their motion, and the relaxation times, and the period of time required for the nuclei to return to their original states in the main, static magnetic field after being subjected to a brief additional magnetic field. These relaxation times reflect the physical-chemical properties of tissue and the molecular environment of its hydrogen nuclei. Only hydrogen atoms are present in human tissues in sufficient concentration for current use in clinical MRI.
Magnetic Resonance Angiography (MRA) is a non-invasive diagnostic test that is an application of MRI. By analyzing the amount of energy released from tissues exposed to a strong magnetic field, MRA provides images of normal and diseased blood vessels, as well as visualization and quantification of blood flow through these vessels.
2. General Clinical Utility
Overall, MRI is a useful diagnostic imaging modality that is capable of demonstrating a wide variety of soft-tissue lesions with contrast resolution equal or superior to CT scanning in various parts of the body.
Among the advantages of MRI are the absence of ionizing radiation and the ability to achieve high levels of tissue contrast resolution without injected iodinated radiological contrast agents. Recent advances in technology have resulted in development and Food and Drug Administration (FDA) approval of new paramagnetic contrast agents for MRI which allow even better visualization in some instances. Multi-slice imaging and the ability to image in multiple planes, especially sagittal and coronal, have provided flexibility not easily available with other modalities. Because cortical (outer layer) bone and metallic prostheses do not cause distortion of MR images, it has been possible to visualize certain lesions and body regions with greater certainty than has been possible with CT. The use of MRI on certain soft tissue structures for the purpose of detecting disruptive, neoplastic, degenerative, or inflammatory lesions has now become established in medical practice.
Phase contrast (PC) and time-of-flight (TOF) are some of the available MRA techniques at the time these instructions are being issued. PC measures the difference between the phases of proton spins in tissue and blood and measures both the venous and arterial blood flow at any point in the cardiac cycle. TOF measures the difference between the amount of magnetization of tissue and blood and provides information on the structure of blood vessels, thus indirectly indicating blood flow. Two-dimensional (2D) and three dimensional (3D) images can be obtained using each method.
Contrast-enhanced MRA (CE-MRA) involves blood flow imaging after the patient receives an intravenous injection of a contrast agent. Gadolinium, a non-ionic element, is the foundation of all contrast agents currently in use. Gadolinium affects the way in which tissues respond to magnetization, resulting in better visualization of structures when compared to un-enhanced studies. Unlike ionic (i.e., iodine-based) contrast agents used in conventional contrast angiography (CA), allergic reactions to gadolinium are extremely rare. Additionally, gadolinium does not cause the kidney failure occasionally seen with ionic contrast agents. Digital subtraction angiography (DSA) is a computer-augmented form of CA that obtains digital blood flow images as contrast agent courses through a blood vessel. The computer "subtracts" bone and other tissue from the image, thereby improving visualization of blood vessels. Physicians elect to use a specific MRA or CA technique based upon clinical information from each patient.
B. Nationally Covered MRI and MRA Indications
1. MRI
Although several uses of MRI are still considered investigational and some uses are clearly contraindicated (see subsection C), MRI is considered medically efficacious for a number of uses. Use the following descriptions as general guidelines or examples of what may be considered covered rather than as a restrictive list of specific covered indications. Coverage is limited to MRI units that have received FDA premarket approval, and such units must be operated within the parameters specified by the approval. In addition, the services must be reasonable and necessary for the diagnosis or treatment of the specific patient involved.
- MRI is useful in examining the head, central nervous system, and spine. Multiple sclerosis can be diagnosed with MRI and the contents of the posterior fossa are visible. The inherent tissue contrast resolution of MRI makes it an appropriate standard diagnostic modality for general neuroradiology.
- MRI can assist in the differential diagnosis of mediastinal and retroperitoneal masses, including abnormalities of the large vessels such as aneurysms and dissection. When a clinical need exists to visualize the parenchyma of solid organs to detect anatomic disruption or neoplasia, this can be accomplished in the liver, urogenital system, adrenals, and pelvic organs without the use of radiological contrast materials. When MRI is considered reasonable and necessary, the use of paramagnetic contrast materials may be covered as part of the study. MRI may also be used to detect and stage pelvic and retroperitoneal neoplasms and to evaluate disorders of cancellous bone and soft tissues. It may also be used in the detection of pericardial thickening. Primary and secondary bone neoplasm and aseptic necrosis can be detected at an early stage and monitored with MRI. Patients with metallic prostheses, especially of the hip, can be imaged in order to detect the early stages of infection of the bone to which the prosthesis is attached.
- MRI may also be covered to diagnose disc disease without regard to whether radiological imaging has been tried first to diagnose the problem.
- MRI with gating devices and surface coils, and gating devices that eliminate distorted images caused by cardiac and respiratory movement cycles are now considered state of the art techniques and may be covered. Surface and other specialty coils may also be covered, as they are used routinely for high resolution imaging where small limited regions of the body are studied. They produce high signal-to-noise ratios resulting in images of enhanced anatomic detail.
2. MRA (MRI for Blood Flow)
Currently covered indications include using MRA for specific conditions to evaluate flow in internal carotid vessels of the head and neck, peripheral arteries of lower extremities, abdomen and pelvis, and the chest. Coverage is limited to MRA units that have received FDA premarket approval, and such units must be operated within the parameters specified by the approval. In addition, the services must be reasonable and necessary for the diagnosis or treatment of the specific patient involved.
Head and Neck
Studies have proven that MRA is effective for evaluating flow in internal carotid vessels of the head and neck. However, not all potential applications of MRA have been shown to be reasonable and necessary. All of the following criteria must apply in order for Medicare to provide coverage for MRA of the head and neck:
MRA is used to evaluate the carotid arteries, the circle of Willis, the anterior, middle or posterior cerebral arteries, the vertebral or basilar arteries or the venous sinuses;
MRA is performed on patients with conditions of the head and neck for which surgery is anticipated and may be found to be appropriate based on the MRA. These conditions include, but are not limited to, tumor, aneurysms, vascular malformations, vascular occlusion or thrombosis. Within this broad category of disorders, medical necessity is the underlying determinant of the need for an MRA in specific diseases. The medical records should clearly justify and demonstrate the existence of medical necessity; and
MRA and CA are not expected to be performed on the same patient for diagnostic purposes prior to the application of anticipated therapy. Only one of these tests will be covered routinely unless the physician can demonstrate the medical need to perform both tests.
Peripheral Arteries of Lower Extremities
Studies have proven that MRA of peripheral arteries is useful in determining the presence and extent of peripheral vascular disease in lower extremities. This procedure is non-invasive and has been shown to find occult vessels in some patients for which those vessels were not apparent when contrast angiography (CA) was performed. Medicare will cover either MRA or CA to evaluate peripheral arteries of the lower extremities. However, both MRA and CA may be useful in some cases, such as:
A patient has had CA and this test was unable to identify a viable run-off vessel for bypass. When exploratory surgery is not believed to be a reasonable medical course of action for this patient, MRA may be performed to identify the viable runoff vessel; or
A patient has had MRA, but the results are inconclusive.
Abdomen and Pelvis
i. Pre-operative Evaluation of Patients Undergoing Elective Abdominal Aortic Aneurysm (AAA) Repair
MRA is covered for pre-operative evaluation of patients undergoing elective AAA repair if the scientific evidence reveals MRA is considered comparable to CA in determining the extent of AAA, as well as in evaluating aortoiliac occlusion disease and renal artery pathology that may be necessary in the surgical planning of AAA repair. These studies also reveal that MRA could provide a net benefit to the patient. If preoperative CA is avoided, then patients are not exposed to the risks associated with invasive procedures, contrast media, end-organ damage, or arterial injury.
ii. Imaging the Renal Arteries and the Aortoiliac Arteries in the Absence of AAA or Aortic Dissection
MRA coverage is expanded to include imaging the renal arteries and the aortoiliac arteries in the absence of AAA or aortic dissection. MRA should be obtained in those circumstances in which using MRA is expected to avoid obtaining CA, when physician history, physical examination, and standard assessment tools provide insufficient information for patient management, and obtaining an MRA has a high probability of positively affecting patient management. However, CA may be ordered after obtaining the results of an MRA in those rare instances where medical necessity is demonstrated.
Chest
i. Diagnosis of Pulmonary Embolism
Current scientific data has shown that diagnostic pulmonary MRAs are improving due to recent developments such as faster imaging capabilities and gadolinium-enhancement. However, these advances in MRA are not significant enough to warrant replacement of pulmonary angiography in the diagnosis of pulmonary embolism for patients who have no contraindication to receiving intravenous iodinated contrast material. Patients who are allergic to iodinated contrast material face a high risk of developing complications if they undergo pulmonary angiography or computed tomography angiography. Therefore, Medicare will cover MRA of the chest for diagnosing a suspected pulmonary embolism when it is contraindicated for the patient to receive intravascular iodinated contrast material.
ii. Evaluation of Thoracic Aortic Dissection and Aneurysm
Studies have shown that MRA of the chest has a high level of diagnostic accuracy for pre-operative and post-operative evaluation of aortic dissection of aneurysm. Depending on the clinical presentation, MRA may be used as an alternative to other non-invasive imaging technologies, such as transesophageal echocardiography and CT. Generally, Medicare will provide coverage only for MRA or for CA when used as a diagnostic test. However, if both MRA and CA of the chest are used, the physician must demonstrate the medical need for performing these tests.
While the intent of this policy is to provide reimbursement for either RA or CA, the Centers for Medicare & Medicaid Services (CMS) is also allowing flexibility for physicians to make appropriate decisions concerning the use of these tests based on the needs of individual patients. CMS anticipates, however, low utilization of the combined use of MRA and CA. As a result, CMS encourages the Medicare Administrative Contractors (MACs) to monitor the use of these tests and, where indicated, require evidence of the need to perform both MRA and CA.
3. MRI for Patients with an Implanted Pacemaker, Implantable Cardioverter Defibrillator, Cardiac Resynchronization Therapy Pacemaker, or Cardiac Resynchronization Therapy Defibrillator
- A MRI is covered when used according to the FDA labeling in an MRI environment for patients with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator.
- Any MRI for patients with an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator that does not have FDA labeling specific to use in an MRI environment is only covered under the following conditions:
- MRI field strength is ≤ 1.5 Tesla;
- It has been ≥ 6 weeks since a patient’s device implantation or any lead revision or surgical modification;
- The patient is not pacemaker-dependent;
- The implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator system has no fractured, epicardial, or abandoned leads;
- The facility has implemented a checklist which includes the following:
- patient assessment is performed to identify the presence of an implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator;
- before the scan benefits and harms of the MRI scan are communicated with the patient or the patient’s delegated decision-maker;
- prior to the MRI scan, the implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator is interrogated and programmed into the appropriate MRI scanning mode;
- a qualified physician, nurse practitioner or physician assistant with expertise with implanted pacemakers, implantable cardioverter defibrillators, cardiac resynchronization therapy pacemakers, or cardiac resynchronization therapy defibrillators must directly supervise;
- a discharge plan that includes before being discharged from the hospital/facility, the patient is evaluated and the implanted pacemaker, implantable cardioverter defibrillator, cardiac resynchronization therapy pacemaker, or cardiac resynchronization therapy defibrillator reinterrogated to detect and correct any abnormalities that might have developed during the MRI.
B. Contraindications and Nationally Non-Covered Indications
- Contraindications
The MRI is not covered when the following patient-specific contraindications are present:
- MRI during a viable pregnancy.
- The danger inherent in bringing ferromagnetic materials within range of MRI units generally constrains the use of MRI on acutely ill patients requiring life support systems and monitoring devices that employ ferromagnetic materials.
- The long imaging time and the enclosed position of the patient may result in claustrophobia, making patients who have a history of claustrophobia unsuitable candidates for MRI procedures.
- Nationally Non-Covered Indications
CMS has determined that MRI of cortical bone and calcifications, and procedures involving spatial resolution of bone and calcifications, are not considered reasonable and necessary indications within the meaning of section 1862(a)(1)(A) of the Act, and are therefore non-covered.
MRI is not covered for patients with metallic clips on vascular aneurysms.
C. Other
All other uses of MRI or MRA for which CMS has not specifically indicated coverage or non-coverage continue to be eligible for coverage through individual MAC discretion.


